IL-2 is a critical T cell cytokine that promotes effector T cell expansion and survival and improves antitumor functions, but as an immunotherapy, its use has been very limited due to induction of suppressive T cell responses and potentially severe toxicities. In an effort to circumvent IL-2’s less desirable effects, Sultan et al. and Sockolosky et al. have independently come up with new modifications to IL-2 administration, published in Cancer Immunology Research and Science, respectively.
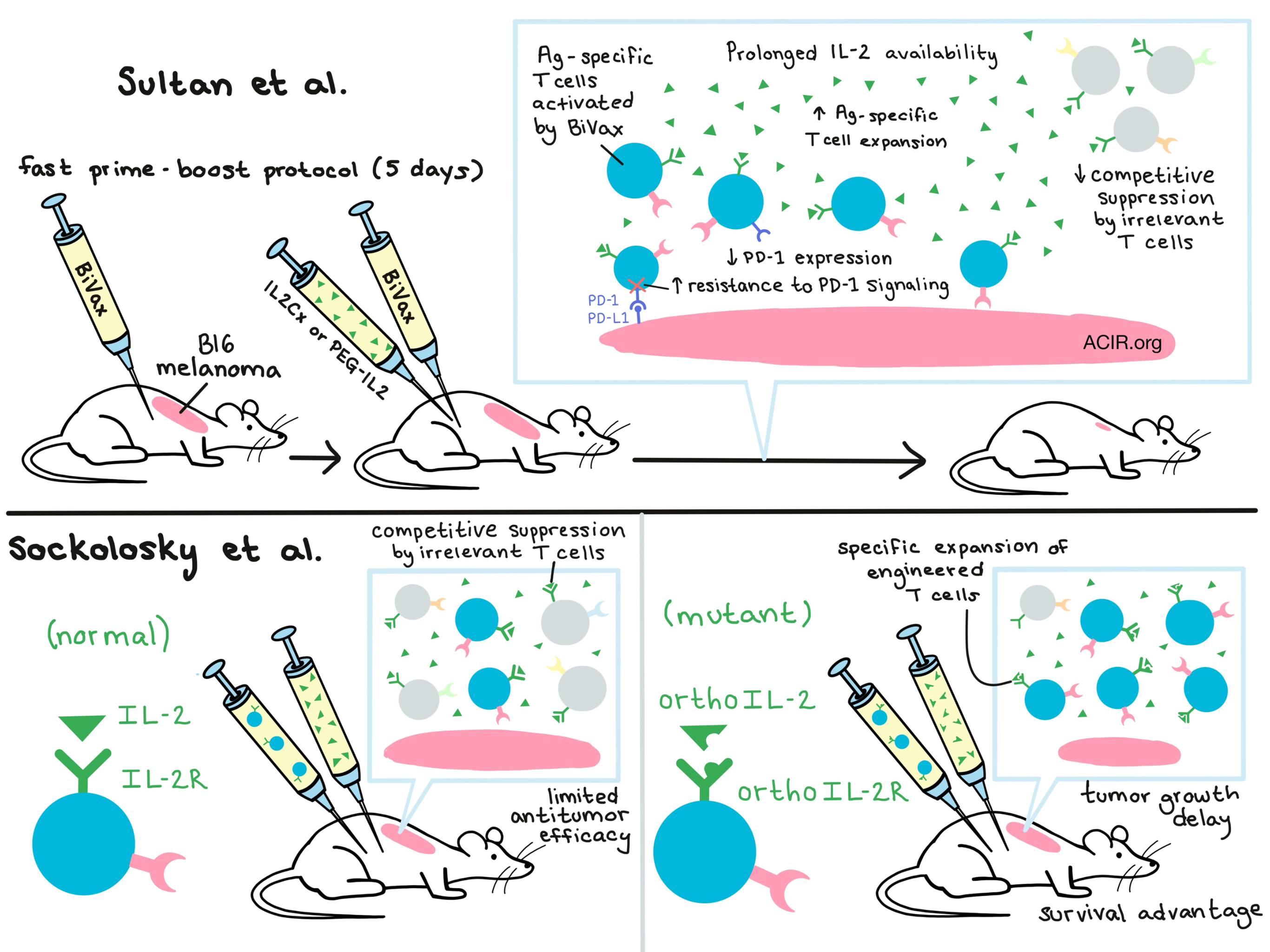
BiVax, a previously developed peptide antigen (Ag) vaccine + poly-IC adjuvant combination developed by Sultan et al., can generate significant Ag-specific T cell responses against defined tumor antigens, but requires co-treatment with anti-PD-1 for optimal antitumor response. Based on their observations that expansion of Ag-specific T cells can be limited by competition with irrelevant T cells, the team evaluated the addition of IL-2 in the form of IL-2/anti-IL-2 complexes (IL2Cx) after vaccination and achieved enhanced Ag-specific T cell expansion in vivo. Interestingly, they found that IL2Cx administration enhanced the vaccine-induced antitumor effect only when it was administered with the vaccine boost (at least 5 days after the vaccine prime), indicating that Ag-specific T cells may need time to become sufficiently activated and upregulate IL-2 receptor expression before they can be prompted to proliferate.
With the proper timing, the addition of IL2Cx to BiVax improved the antitumor efficacy of Ag-specific T cells against established subcutaneous B16 melanoma, delaying tumor growth and extending overall survival compared to controls. Expectedly, the researchers found that the antitumor efficacy was due in part to the increased number of Ag-specific T cells, but they also noted that the IL2Cx treatment reduced expression of PD-1 on the stimulated T cells. Surprisingly, by comparison to a similar vaccine formulation, they came to realize that neither cell number nor PD-1 expression fully explained the increased antitumor efficacy. Through a series of experiments, the researchers found that IL2Cx also enhanced the ability of T cells to resist PD-1-induced negative signaling via increased expression of pSTAT5; this allowed the cells to remain functional even in tumor microenvironments expressing high levels of PD-L1 and PD-L2.
Next, the researchers demonstrated that the antibody/cytokine complex enhanced vaccine efficacy by extending the in vivo half-life of available IL-2 in the serum rather than by antibody-mediated targeting to IL-2 receptors. In this vein, they went on to show that half-life-extended pegylated IL-2 (PEG-IL2)was similarly effective. Mice treated with Bi-Vax + IL2Cx or PEG-IL2 were not cured and eventually developed tumors despite persistence of Ag-specific T cells and continued tumor expression of the antigen, suggesting that extended IL2Cx dosing might be required, although other explanations are possible.
In a separate study focusing on how to use IL-2 to improve expansion of engineered T cells, Sockolosky et al. created IL-2 cytokine-receptor orthogonal pairs that interact preferentially with one another, but not with their natural cytokine and receptor counterparts. To develop the pairs, the researchers focused their efforts on the interaction between IL-2 and the IL-2Rβ chain; of the three IL-2R chains, IL-2Rβ is required for signal transduction and can bind to IL-2 independently. Based on the known structure of the IL-2/IL-2R complex, the researchers generated a double mutant orthoIL-2Rβ incapable of binding wild-type IL-2. They then used yeast display to evolve IL-2 variants capable of binding to orthoIL-2Rβ but not wild-type IL-2Rβ. This yielded two standout orthoIL-2 mutant variants, 1G12 and 3A10.
To test their system in vivo, the researchers engineered T cells to express the mutant orthoIL-2Rβ and adoptively transferred them into immunocompetent mice. Using the orthoIL-2, they were able to direct IL-2 signaling to preferentially expand adoptively transferred T cells, with the 3A10 variant being more selective but slightly less potent, and the 1G12 variant being the opposite. Fusing each of the orthoIL-2 variants to albumin extended in vivo half-life; this increased the activity of both variants, but only the orthoIL-2 3A10 variant retained strong target specificity. Ex vivo evaluation of orthoIL-2Rβ T cells that had been expanded in vivo with orthoIL-2 showed that they produced significantly more IFNγ than wild-type T cells expanded with IL-2. While PD-1 levels were comparable between these groups, TIM-3 exhaustion was significantly lower on orthoIL-2Rβ T cells. This suggested that there may be additional off-target effects of IL-2 not induced with orthoIL-2.
Finally, to test clinical relevance, Sockolosky et al. engineered T cells with a tumor-specific TCR and orthoIL-2Rβ and adoptively transferred them into mice with B16-F10 melanoma. Administration of orthoIL-2 3A10 (or 1G12 at a dose that had minimal off-target activity on wild-type IL-2R) produced a significant tumor growth delay and survival advantage, comparable to the advantage produced with standard high-dose IL-2. Overall, Sockolosky et al. conclude that this approach could be used to redirect the specificity of IL-2 toward engineered T cells, enabling the selective expansion of only desired subsets in adoptive cell therapy with reduced off-target activity and toxicity. This strategy works on other cells expressing the common IL-2 γ-chain (such as Tregs or B cells) and so could be broadly useful as both a research tool and in the clinical setting.
by Lauren Hitchings



