Glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) signaling stimulates T cell activation and proliferation and can inhibit suppressive effects of regulatory T cells (Treg). However, agonistic GITR antibodies have shown limited results in the clinic, which may be due to limited receptor clustering-mediated signaling of this TNFR superfamily member. Therefore, Chan and Belmar et al. developed a protein engineering approach to improve GITR clustering and efficacy. Their results were recently published in Nature Cancer.
The human GITR ligand (GITR-L):GITR complex was crystalized and shown to consist of a GITR-L homotrimer, with each GITR-L monomeric subunit binding to a homodimer of GITR receptors. The authors then assessed the role of GITR clustering by building a series of GITR-L- or GITR agonist domain-containing constructs, and evaluated the effects of different levels of receptor clustering on T cell costimulation. When higher numbers of GITR-L were included in the constructs, there was more costimulation, consistent with an avidity-based clustering effect. The highest activity was achieved by a hexameric ligand–trimer fusion molecule (12 GITR-L trimers arrayed by hexamerizing Fc domains).
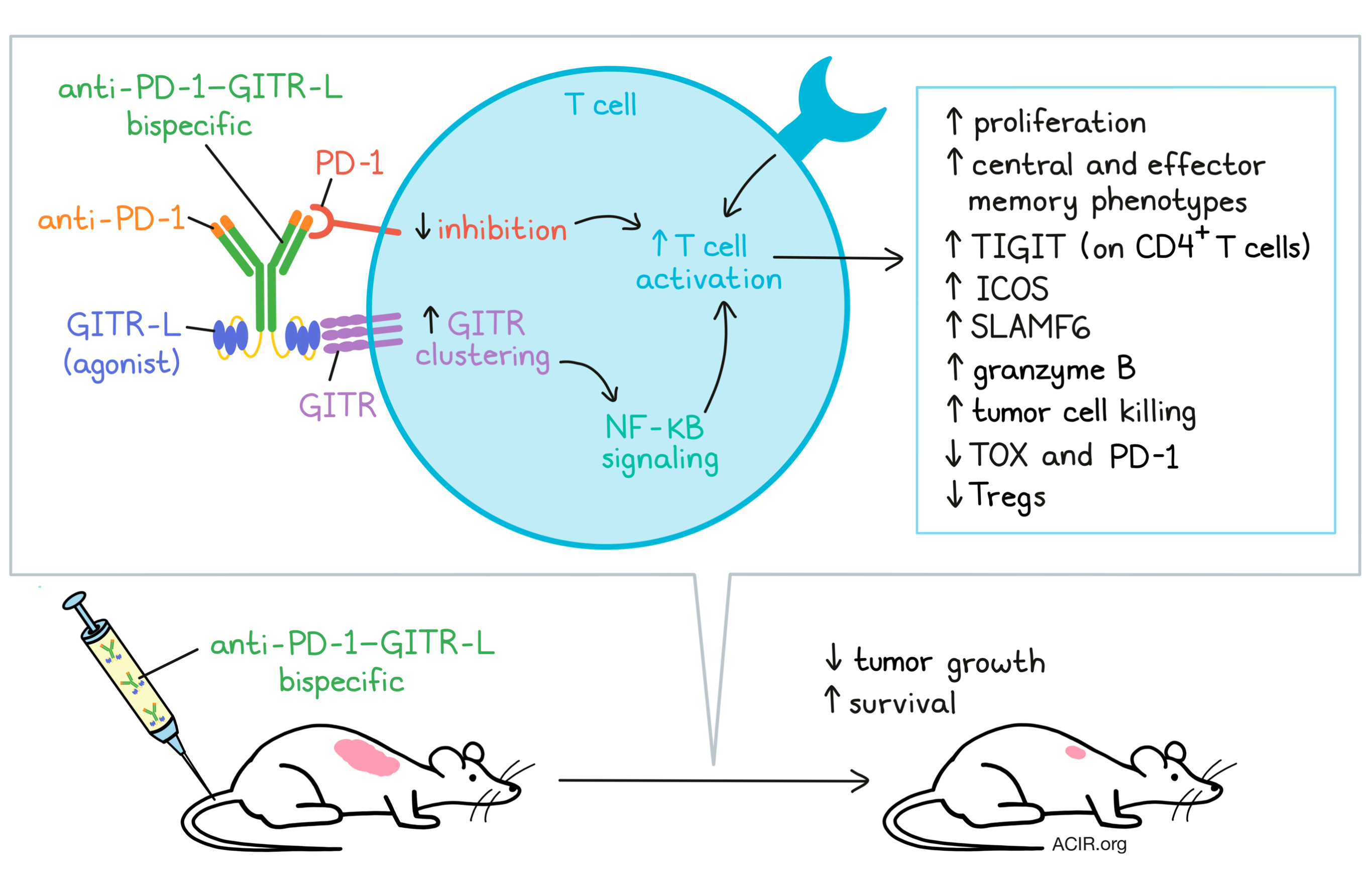
To target GITR stimulation to activated T cells, Chan and Belmar et al. designed an anti-PD-1–GITR-L bispecific, in which anti-PD-1 is used to enhance GITR clustering and NF-κB signaling of T cells. The bispecific is an anti-huPD-1 antibody fused on its C-terminus, via flexible (Gly3Ser)4 linkers, to the N terminus of the extracellular domains of two human GITR-L trimers, each connected by (Gly3Ser)4 linkers.
The researchers confirmed that the targets PD-1 and GITR are expressed on peripheral T cells and tumor-infiltrating lymphocytes (TILs) from various cancer types. They found a high correlation between GITR and PD-1 gene expression in T cells from head and neck squamous cell carcinoma. Spatial distribution imaging analysis showed that some CD8+PD-1+ T cells expressed GITR in tumor, stromal, and tumor-proximal lymph node regions in both inflamed and excluded tumors.
Using a surrogate mouse version of their bispecific (anti-muPD-1–muGITR-L), the researchers assessed its activity and mechanism of action. There was a dose-dependent antitumor effect after one dose in mice bearing s.c. syngeneic CT26 or EMT6 tumors. Treatment induced a dose-dependent increase in the fraction of TIGIT+ CD4+ T cells, CD62L+CD44+ CD4+ central memory T cells, and Ki67+ CD4+ and CD8+ T cells in the periphery. In tumor-draining lymph nodes (TDLN), the percentage of ICOS+, CD62L-CD44+ effector memory, and KI67+ cells within CD4+ and CD8+ T cell populations increased, while TILs contained an increased proportion of Ki67+ CD8+ T cells, more granzyme B (GZMB), and a decreased proportion of Tregs.
In the CT26 model, tumors contained more CD8+ effector T cells and NK cells. Immune cell gene marker analyses of single-cell mRNAseq data also revealed an increase in CD8+ T and NK cells; higher levels of various genes involved in the activation, survival, and homeostasis of T cells; and an increase in interferon and cytokine-related signaling pathways. In addition, in CD8+ T cells, there was an increase in the progenitor marker SLAMF6 and a decrease in the exhaustion markers TOX and PD-1. No liver toxicity or acute cytokine release syndrome was observed after treatment.
Moving to humanized target knock-in (KI) mouse models with either human GITR or PD-1, two chimeric bispecifics were tested, namely anti-muPD-1–huGITR-L and anti-huPD-1–muGITR-L. The bispecifics inhibited the growth rate of MC38 tumors in the corresponding mice, similar to effects seen with the surrogate anti-muPD-1–muGITR-L in WT mice. However, no efficacy was detected in WT mice that received the chimeric bispecifics, which confirms that co-engagement of PD-1 and GITR is necessary for its activity. In the humanized mice, the number and phenotypes of effector and memory T cells were comparable to those observed in WT mice treated with the fully murine bispecifics.
In NSG mice with PC-3 or HCT-116 tumors and engrafted with allogeneic human T cells and monocyte-derived dendritic cells, a single dose of anti-huPD-1–huGITR-L inhibited tumor growth, even when it was given after tumors were established.
The effects of anti-muPD-1–muGITR-L on tumor growth inhibition were different from those of the monotherapies used individually or in combination. It induced longer overall survival in CT26, EMT6, and JC tumor models, and efficacy was correlated with an increase in the percentage of ICOS- and Ki67-expressing CD8+ T cells. Furthermore, the bispecific induced memory responses, as multiple CT26 tumor rechallenges were rejected in long-term survivors. Further, tetramer staining revealed the presence of the CT26-endogenous MuLV-gp70 antigen-specific T cells. CD8+ T cells from TDLNs showed an increase in the number of antigen-specific granzyme B+ T cells, as well as more tumor killing compared to those from mice treated with the combination strategy. The granzyme B was mainly produced by CD8+ T cells, not NK cells, and when CD8+ T cells were depleted, the efficacy was absent. These data suggest that the mode of action of the bispecific is through activation of CD8+ T cells.
NanoString gene expression analysis also revealed differences between treatment with anti-muPD-1–muGITR-L and treatment with the monotherapies or their combination. Upregulated gene signatures included those associated with adaptive and innate immune responses and cytotoxicity. There was an increase in CD8a, GZMB, and genes related to activating and inhibitory receptor pathways on NK cells. Furthermore, upregulation of genes involved in cellular responses to IFNγ and MAP kinases, chemokine-mediated pathways, lymphocyte chemotaxis, and immune/inflammatory responses were detected.
Finally, the researchers moved to experiments with non-human primates. Anti-huPD-1–huGITR-L was shown to bind monkey PD-1 and GITR on PBMCs with similar affinity to what has been observed in human PBMCs, and to induce similar signal transduction signaling. Treatment of monkeys resulted in a dose-dependent increase in TIGIT on CD4+ Tcm cells and an increase in Ki67 on CD4+CD45RA- memory T cells. No significant changes in cytokine and chemokine plasma levels were observed, suggesting safety. There was a correlation between target saturation and modulation of pharmacodynamic effects, suggesting a similar mode of action as found in the mice.
These data suggest that anti-huPD-1–huGITR-L may be more effective than combined PD-1 inhibition and GITR agonism due to improved GITR clustering, resulting in stimulation of tumor-specific T cell responses. Improving the efficacy of co-stimulatory molecule agonism could be an important step to treatment for tumors that respond poorly to checkpoint blockade.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, several researchers on the team answered our questions.

What prompted you to tackle this research question?
Multiple studies in mice have demonstrated that GITR ligation by an agonistic antibody or a GITR–ligand construct induces an immunological response and increases resistance to tumors by accumulation of CD8+ T cells and depletion of intratumoral Tregs. However, anti-GITR antibodies show a limited therapeutic effect in several human clinical trials, even though they induce a reduction of circulating and intratumoral Tregs. These results suggest that anti-GITR-mediated Treg cell depletion is not sufficient for inducing survival in humans, and that their limited bioactivity is potentially due to a lack of T cell activation and proliferation mediated by suboptimal GITR receptor clustering.
What was the most surprising finding of this study for you?
The anti-PD-1–GITR-L bispecific was designed to optimize GITR clustering and NF-κB signaling of primed antigen-specific T cells in cis. This idea was inspired by three critical findings. First, the x-ray structure of the human GITR-L–GITR complex reveals a non-covalent GITR-L-mediated receptor homodimerization, which represents one of multiple states for optimal GITR receptor clustering. Second, optimal receptor oligomerization and bioactivity was demonstrated by using engineered hexameric GITR-L- and anti-GITR-based constructs. And third, the anti-PD-1–GITR-L bispecific induced a significant increase in NF-κB signaling using PD-1+GITR+ Jurkat reporter cells due to anti-PD-1-dependent GITR clustering, which is independent of FcγR-mediated T cell activation. The anti-PD-1–GITR-L bispecific represents a different approach for T cell agonism for cancer immunotherapy that can be applied to different members of the tumor necrosis factor receptor superfamily (TNFRSF).
What was the coolest thing you’ve learned (about) recently outside of work?
The recent pandemic situation has disrupted everyone’s life, including scientists, causing a good many of them to think about acquiring new skills. Our team learned how to enjoy spending time reading, painting, cooking, running, and dancing though Zoom.



