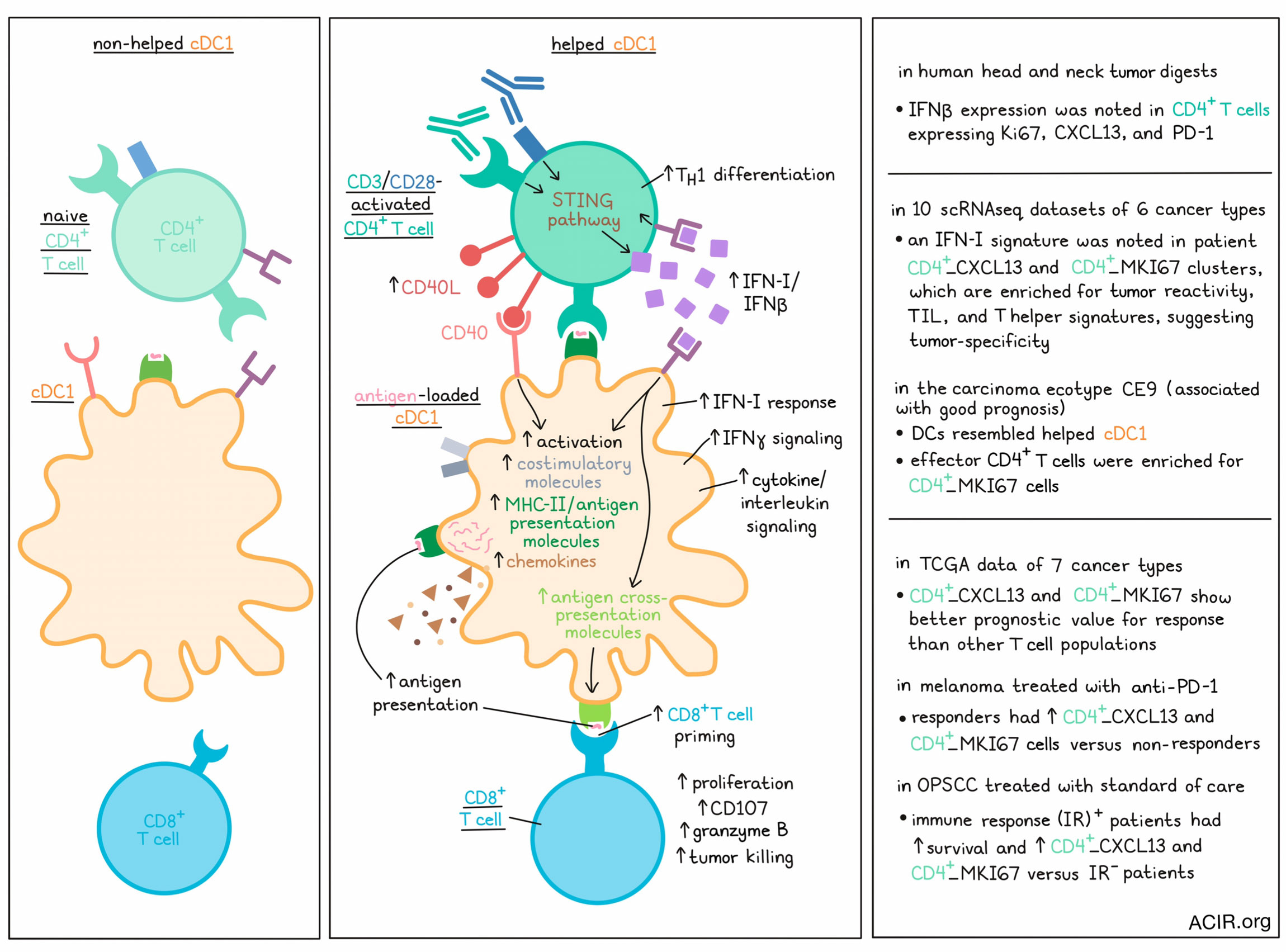
CD4+ T helper cells can stimulate CD8+ T cell responses by interacting with dendritic cells (DCs) through a process termed “DC licensing”. The conventional type 1 DC (cDC1) subset has been identified as the essential DC subtype for this process. Lei et al. investigated the mechanism of cDC1 licensing in human cells and the tumor microenvironment (TME) setting, and their data were recently published in Cellular and Molecular Immunology.
To assess the characteristics of “helped” and “non-helped” cDC1s, the researchers performed co-culturing assays in which human peripheral cDC1s were cocultured with naive (non-helped) or CD3/CD28-activated CD4+ T cells (helped). Gene set enrichment analysis revealed that in the helped cDC1s, cytokine/interleukin signaling, IFN-I response, and IFNγ signaling were upregulated. The researchers hypothesized that CD4+ T cells produced IFN-I in this setting. To test this, cDC1s were stimulated with activated CD4+ T cells or Poly(I:C). Expression of IFNα and IFNβ was upregulated in cDC1s stimulated by Poly(I:C), but not in those stimulated by activated CD4+ T cells. Additionally, IFNβ, but not IFNα, was upregulated in CD4+ T cells stimulated with both CD3 and CD28. Both results were consistent with CD4+ T cells being the source of IFN-I.
In myeloid cells, the STING pathway induces IFN-I production. STING signaling markers were measured in activated CD4+ T cells to determine whether their IFN-I production was also related to this pathway. Increased expression of STING pathway markers pSTING, pTBK1, and pIRF3 were detected at 24 hours after stimulation, and by 48 hours, IFNβ was produced. Using the STING agonist 2’3’-cGAMP in the assay showed increased protein levels of IFNβ, pSTING, and pTBK1, as well as IFN-I-stimulated gene products in the T cells. Furthermore, the agonist induced expression of markers of Th1 differentiation. STING inhibition, on the other hand, decreased expression of IFNβ in the T cells.
Lei et al. then determined whether cDC1s respond to IFN-I produced by CD4+ T cells by repeating the assay in the presence or absence of an anti-IFNα/β receptor 2 (IFNAR2)-blocking antibody. cDC1s upregulated expression of costimulatory molecules, chemokines, and antigen presentation pathway components after culture with IFN-I or after co-culturing with activated CD4+ T cells, which did not occur when IFNAR2 was blocked.
The researchers assessed whether IFNβ produced by CD4+ T cells impacted cDC1-mediated CTL responses in humans using an in vitro tumor antigen-specific CTL priming platform. CD8+ T cells were retrovirally transduced to express an HLA-A2-restricted MART-126-35 peptide-specific TCR. cDC1s were loaded with MART-115-40 long peptide or dead MART-1-expressing Mel526 cell debris to create antigen cross-presentation, and these cells were then exposed to activated MART-1 CD4+ T cells pretreated with or without IFNβ siRNA. Following long peptide loading of cDC1s, the proliferation of tetramer-positive CD8+ T cells did not differ between the siRNA-transfected and control CD4+ T cells, but granzyme B production was lower in the IFNβ1 siRNA-transfected setting. Loading of dead cells though resulted in a reduction of both proliferation and granzyme B when CD4+ T cells were treated with IFNβ siRNA, indicating that utilization of this natural antigen source was more sensitive to IFNβ stimulation from CD4+ T cells.
Activated CD4+ T cells upregulate the expression of CD40L, which can interact with CD40 on cDC1 for cDC1 licensing. To determine the contributions of IFNβ and CD40L in CD4+ T cell-mediated help, CD4+ T cells were edited by CRISPR-Cas9 to be deficient in IFNβ and/or CD40L, and were used in the activation coculture assay. Loss of IFNβ or CD40L in activated CD4+ T cells reduced expression of activation markers, MHC class II, and chemokines in cDC1s. However, only the IFNβ-deficient CD4+ T cells reduced the ability to upregulate key molecules involved in antigen cross-presentation, suggesting a role of IFN-I signaling in cDC1 licensing that is CD40-independent. In the CTL assay, loss of IFNβ or CD40L in activated CD4+ T cells reduced the priming of antigen-specific CD8+ T cells. CTL tumor cell killing capacity was also higher in cultures primed by helped cDC1s. In cultures primed with CD4+ T cells that were IFNβ- or CD40L-deficient, granzyme B production and CD107a expression decreased, and these CD8+ T cells were less capable of killing tumor cells.
Lei et al. then investigated the role of IFN-I-producing CD4+ T cells in cDC1 licensing in the human TME. In head and neck cancer tissue digests, IFNβ was detected in clusters of CD4+ T cells expressing Ki67, CXCL13, and PD-1. A cohort of ten scRNAseq datasets, including six types of human cancer, revealed that genes related to the IFN-I signature were highest expressed in the CD4+_ISG15, CD4+_CXCL13, and CD4+_MKI67 clusters. Gene signatures previously related to tumor reactivity in tumor-infiltrating lymphocytes (TIL) were enriched in the CD4+_CXCL13 and CD4+_MKI67 clusters, suggesting that the IFN-I-producing CD4+ T cells in the TME might be tumor-specific. A previous publication showed that a tumor-specific CXCL13+CD4+ T helper cell population in the TME interacts with LAMP3+ DCs, and the gene signature of this CXCL13+ T helper population was also enriched in the CD4+_CXCL13 and CD4+_MKI67 clusters. Another previous study detected various carcinoma ecotypes (CE), of which CE9 is correlated with good prognosis and is characterized by the combined presence of DCs, effector CD4+ and CD8+ T cells. The authors previously showed that the tumor-infiltrating DC signature in CE9 shares many characteristics with the “helped” cDC1 signature. Here, they detected that the gene expression signature of CD4+ T cells in CE9 is highly enriched in the detected CD4+_MKI67 population.
To determine the clinical relevance of these findings, the researchers performed survival analyses on TCGA data of seven cancer histologies. The CD4+_CXCL13 and CD4+_MKI67 signatures had a better prognostic value than other CD4+ T cell populations. Furthermore, to assess the predictive value of these populations for response to therapy, a melanoma cohort receiving anti-PD-1 therapy and an HPV+ oropharyngeal squamous cell carcinoma (OPSCC) cohort receiving standard-of-care therapies were analyzed. In melanoma, responders had higher CD4+_MKI67 and CD4+_CXCL13 signature levels than nonresponders. In OPSCC, when splitting patient groups based on immune response-positive (IR+) and -negative based on the presence of tumor-specific T cells among TILs, the IR+ patients had improved survival and higher levels of the CD4+_MKI67 and CD4_CXCL13 populations.
Together, these data suggest that IFN-I signaling induced by activated CD4+ T cells licenses cDC1 to induce antitumor CD8+ T cell responses, which might be associated with improved clinical outcomes. This helper role of CD4+ T cells in the TME may help overcome suboptimal priming of CD8+ T cells, and further research could shine more light on the role of various immunotherapies in improving this helper role and overcoming inhibition of this mechanism in the TME.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, lead author Yanling Xiao answered our questions.

What was the most surprising finding of this study for you?
In our pursuit to unravel the secrets behind cDC1's exceptional ability in tumor cell-associated antigen presentation, we explored the molecular processes. When activation of the IFN-I pathway occurred in the licensed cDC1, the source of IFN-I surprised us—it wasn't from presumed autocrine signaling, but originated from activated CD4+ T cells. The CD40/CD40L axis has been acknowledged as the primary player in cDC1 licensing. However, our discovery reveals that, despite intact CD40L/CD40 signaling, IFNβ-/- CD4+ T cells are unable to effectively license cDC1. Importantly, we find that MHC-I antigen cross-presentation in licensed cDC1 is mediated by IFN-I signaling rather than CD40. Through extensive exploration of the TME across multiple tumor types, we unveil a clinically favored subpopulation of IFN-I-producing CXCL13+CD4+ T-cells, reported to co-localize with helped/licensed cDC1.
What is the outlook?
Our findings advance cancer immunotherapy, showcasing cDC1 licensing in the human TME of favorable, T cell-infiltrated cancers. Transcriptomic evidence elucidates CD4+ T cell and cDC cross-talk, presenting a coherent scenario of CD4+ T cell help via cDC1 in the TME, mechanistically connecting with the historically known positive association of IFN-I signals. Our research consolidates evidence from recent impactful publications, providing a comprehensive understanding of CD4+ T cell and cDC1 interplay in the TME and paving the way for cancer immunotherapy advancements. Overall, this underscores the continual importance of research in this promising field. For that, we have recently been awarded funding from the Dutch Cancer Society (KWF) to bolster our efforts in identifying new targets for cancer immunotherapy in helped/licensed cDC1 within the TME.
What was the coolest thing you’ve learned (about) recently outside of work?
I am Chinese Dutch and lived in Japan for my Ph.D. Recently, I discovered raw fish roe at a delightful seafood stand in a charming town called Oegstgeest. Missing the Japanese dish called mentaiko, I decided to make it myself. Mentaiko is a delicacy consisting of roe sacs marinated in salt and chili pepper, enjoyed as an accompaniment to rice dishes. The process took seven days, and the final result brought me immense satisfaction, even though I am the sole enthusiast in the family. Delightful culinary creations have a unique ability to bring joy, not only to those who savor them, but also to the individuals who create these delectable dishes. The art of preparing food becomes a source of happiness, and when one has the skill to create dishes that bring profound happiness to many, it is a culinary achievement. The same holds true for a good piece of creative scientific work.



