To improve the response rate to cancer immunotherapies, Wang and Sun et al. sought to identify factors beyond the PD-1 axis and other known checkpoint inhibitors that may affect T cell function. Utilizing a high-throughput screening approach as well as in vitro and in vivo studies, the researchers identified Siglec-15 as a suppressor of T cell function and a potential novel immunotherapy target. The results were recently published in Nature Medicine.
The researchers began by designing a high-throughput, genome-scale T cell activity array (TCAA) to screen and identify cell surface modulators of T cell activity. The TCAA included artificial antigen-presenting cells that were individually transfected with human genes encoding >90% of transmembrane proteins and that were engineered to stimulate the T cell receptor of NF-κB/NFAT reporter Jurkat cells. In addition to the known costimulatory, inhibitory, and apoptosis-inducing regulator genes, Siglec-15 appeared in this screening as a consistent suppressor of T cell activity.
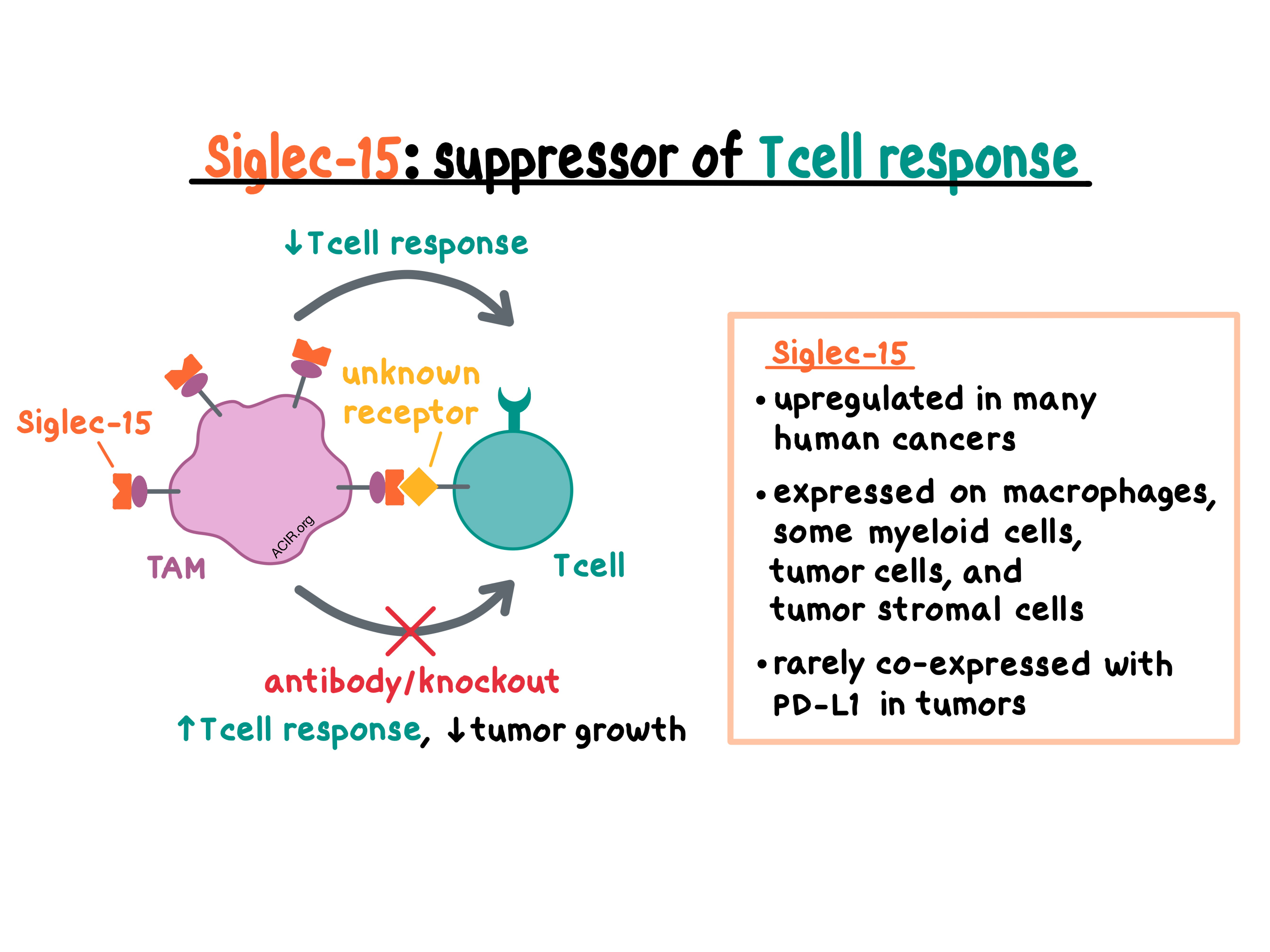
Siglec-15 has previously been identified as one of the sialic acid-binding immunoglobulin-like lectin (Siglec) family members. Normally, Siglec-15 is minimally expressed in human and murine tissues and various cell types, with the exception of macrophages where it is expressed at low levels on the cell surface. The researchers confirmed this by developing and analyzing a Siglec-15-knockout mouse model (S15KO). By comparing S15KO and wild-type mice, the team found that Siglec-15 was expressed at low levels on CD11b+F4/80+ peritoneal macrophages, CD11b+F4/80+ bone marrow-derived macrophages (BMDMs), and CD11b+Gr-1+ myeloid cells, but not on other myeloid or lymphoid cells in wild-type animals.
Next, the researchers examined the effect of Siglec-15 in vitro. Although Siglec-15 was found to have over 30% homology with the B7 gene family, IFNγ induced the expression of B7-H1 (PD-L1), but inhibited the expression of Siglec-15 on mouse myeloid cells and human macrophages, indicating distinct regulation of expression for these related genes. The team then showed that macrophage-expressed Siglec-15 directly suppressed the proliferation and activity of human and murine T cells and inhibited IFNγ and TNFα secretion. It remains unclear which receptor on T cells binds Siglec-15.
Turning to in vivo studies, the researchers utilized the OT-I/OVA system and showed that Siglec-15 suppressed antigen-specific T cell responses mostly by regulating cell growth, but not by inducing apoptosis. In S15KO mice and in mice with selective Siglec-15 knockout on macrophages, T cells proliferated at a faster rate than in wild-type mice – an effect that was associated with decreased IL-10 in sera. Anti-IL-10 boosted the expansion of antigen-specific T cells, abrogating the effect of Siglec-15 in wild-type mice and suggesting that the Siglec-15-mediated suppression relies on IL-10. Overall, these experiments confirmed that Siglec-15 on macrophages suppresses T cell responses in vivo.
Looking in the TCGA expression database, Wang and Sun et al. found that Siglec-15 is upregulated in many human cancers, suggesting potential clinical relevance. Immunohistochemical analysis of 241 NSCLC samples demonstrated that Siglec-15 was found on tumor cells, tumor-associated stromal cells, and tumor-associated CD68+ myeloid cells. Interestingly, consistent with murine in vitro data, there was no correlation between PD-L1 and Siglec-15 expression, with only about 3% of patient cases expressing both markers.
In contrast to human cancers, Siglec-15 was not found in common mouse tumor lines, such as B16, CT26, and MC38. Therefore, the researchers utilized B16-GM-CSF melanoma tumors (which, unlike B16 tumors, are highly infiltrated by tumor-associated macrophages [TAMs]) and GL261 glioma tumors (which are infiltrated by macrophages and microglia) to model and study human tumors with Siglec-15 expression. S15KO mice inoculated with B16-GM-CSF tumors had decreased tumor growth and increased survival compared with wild-type mice, and this was due to an intratumoral expansion of CD8+ T cells, NK cells, and inflammatory myeloid cells, as well as a decrease in TAMs. The antitumor effect was dependent on CD8+ T cells in this model. Similar antitumor effects were observed in the GL261 tumors. Together, these results suggest that Siglec-15 on host macrophages suppresses CD8+ T cells and promotes the growth of tumors in these two models.
To find out if the suppressive effect of Siglec-15 could be therapeutically reversed, the team generated anti-S15, a monoclonal antibody capable of binding both human and murine Siglec-15. In vitro, anti-S15 blocked Siglec-15 and increased the responses of human and murine T cells. In vivo, anti-S15 modestly reduced the growth of established B16-GM-CSF tumors. Anti-S15 had no effect on the growth of MC38 tumors, which demonstrate very limited Siglec-15 expression, but reduced the growth and metastases of MC38 tumors transduced to overexpress Siglec-15. The authors hypothesized that Siglec-15 may be regulating T cells non-redundantly with the PD-1/PD-L1 axis. In mice with CT26 tumors mixed with BMDMs prior to inoculation, anti-PD-1 and anti-S15 had a modest effect on tumor growth as monotherapies, but were much more efficacious in combination, leading to complete regressions in some animals.
Overall, this study demonstrates that the cell surface expression of Siglec-15 on tumor cells or TAMs can impair the antitumor immune response within the tumor microenvironment, and that Siglec-15 is an immunosuppressive regulator that could be therapeutically targeted. A phase I clinical trial is underway to test the effect of a humanized monoclonal antibody (NC318) on Siglec-15 in solid tumors.
by Anna Scherer



