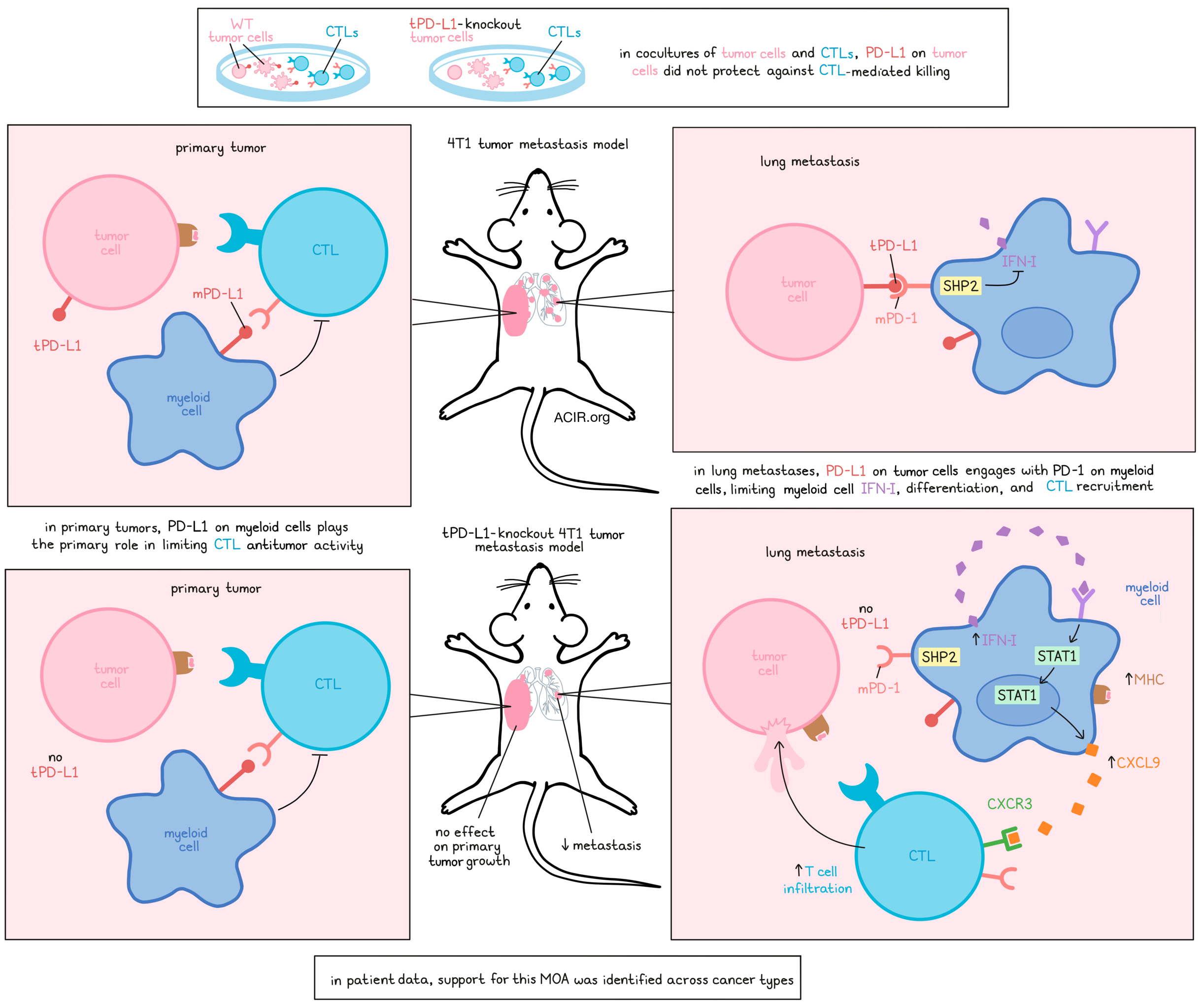
PD-L1 on tumors (tPD-L1) is a well known antagonist of antitumor immune responses, and a biomarker of improved clinical outcomes and survival following PD-1/PD-L1 checkpoint blockade. However, recent research has shown that PD-L1 on other immune cells in the tumor microenvironment may be responsible for suppressing cytotoxic T cells. Taking another look at how tPD-L1 might impact outcomes, Klement and Redd et al. investigated the role of tPD-L1 in metastasis, which accounts for over 90% of mortality in human cancers. Their results, recently published in Cancer Cell, showed that tPD-L1 engages with PD-1 on myeloid cells (mPD-1), which in turn limits cytotoxic T cell recruitment in the metastatic setting.
To begin, the researchers cocultured antigen-specific cytotoxic T lymphocytes (CTLs) with either PD-L1 wild-type (WT) or PD-L1 knockout (KO) tumor cells from various PD-L1+PDL2- tumor cell lines typically resistant to checkpoint blockade. While antigen-specific cytotoxicity was observed in a dose-dependent manner, there was no difference in T cell activation, T cell upregulation of PD-1, cytotoxicity, or killing of target cells between WT and PD-L1 KO tumor cells. Anti-PD-1 also showed no differential effects in these cocultures, suggesting that tPD-L1 does not directly protect tumor cells from CTL attack.
Testing whether the same was true in vivo, Klement and Redd et al. subcutaneously injected WT and PD-L1 KO tumors into mice and showed that both primary tumors grew at the same rate. To study a metastasis model, a small amount of 4T1 cells were orthotopically transplanted into the mammary glands of mice, allowing for the spontaneous development of lung metastases, without the need for primary tumor resection. In this model, WT and PD-L1 KO primary tumors grew at similar rates, but metastases were reduced in mice bearing the PD-L1 KO tumors. Similar results were observed in another experimental lung metastasis model.
Looking at other sources of PD-L1, the researchers identified a population of PD-L1+CD11b+ cells that was increased in primary tumors compared to lung metastases. The administration of anti-PD-1, which blocks engagement by both tumor and immune cells expressing PD-L1, suppressed the growth of established MC38-met tumors, dependent on host PD-1, suggesting that myeloid cell-expressed PD-L1 (mPD-L1) accounts for the observed anti-PD-1-mediated tumor suppression in primary tumors, although the exact inhibitory mechanism of mPD-L1 is still unclear.
Returning to the 4T1 model, Klement and Redd et al. observed increased CD3+ T cells in the metastatic lungs, but not primary tumors, when tPD-L1 was absent. In lymphocyte-deficient mice, WT and PD-L1 KO tumors formed metastases equally. Similarly, genetic ablation of MHC-I or antibody-mediated blockade of CD8+ T cells also equalized metastasis, suggesting that tPD-L1 plays a role in promoting metastatic tumor growth by limiting T cell-mediated tumor control.
Given the differential impact of tPD-L1 on primary versus metastatic tumors, and the absence of direct CTL inhibitory activity observed in vitro, the researchers hypothesized that an additional factor in the lung metastatic niche likely enables tPD-L1 to exert indirect effects on CTLs. Investigating this, they performed RNA sequencing of lungs colonized by WT and PD-L1 KO 4T1 metastases, and identified differentially expressed transcripts involved in antigen presentation (B2m, Cd74, Tap1), IFN response (Stat1, BBst2, Irf2), and CTL recruitment (Cxcl9, Cxcl10, Cxcl11). Further, enhanced type I IFN signaling in the PD-L1 KO setting was identified as the driver of these differences.
To better understand how tPD-L1 suppresses IFN-I signaling in metastatic sites, the researchers performed scRNAseq of CD45+ leukocytes from WT and PD-L1 KO tumor-colonized lungs. Analysis revealed that while PD-1 was mostly expressed on T cells, it was also expressed in a population of macrophages/myeloid cells. Surprisingly, analysis of differentially expressed genes showed that differences in gene expression were associated with transcriptional changes in macrophages, with loss of tPD-L1 resulting in dramatic increases in myeloid expression of Cxcl9 and Cxcl10, which encode chemokines that bind to CXCR3 to recruit T cells. In contrast, minimal transcriptional changes were observed in CTLs.
While only a small portion of myeloid cells expressed PD-1, the researchers identified a central progenitor population that diverged into two branches, differentiated by high or low IFN-I signaling. In mice bearing WT tumors, myeloid cell differentiation was driven towards the low-IFN-I pathway, which showed reduced expression of antigen-presenting machinery and T cell-recruiting chemokines, and was associated with PD-1 signaling via SHP2, implicating myeloid PD-1 as the target of tPD-L1. In PD-L1 KO tumors, more myeloid cells were able to differentiate towards the IFN-I-high group, expressing increased antigen-presenting machinery and T cell-recruiting chemokines.
Through a series of knockout and depletion experiments, Klement and Redd et al. demonstrated that at sites of metastasis, tPD-L1 engages mPD-1, which induces SHP2 signaling that suppresses autocrine IFN-I signaling, directing myeloid cell differentiation. When this mechanism is interrupted, IFN-I signaling increases, leading to increased expression of STAT1, MHC-I, MHC-II, CXCL9, and CXCL10. These changes result in increased infiltration by CTLs, which were essential in limiting the development of metastases. In this way, tPD-L1 supports immune evasion and metastasis by limiting CTLs via PD-1 on myeloid cells.
To evaluate the human relevance of this mechanism, the researchers analyzed patient data. In patients with metastatic basal cell carcinoma who had been treated with PD-1 blockade, a myeloid cluster with high IFN-I signaling was inversely correlated with SHP2 activity. Differentiation towards this cluster was enhanced after PD-1 blockade, leading to increased IFN-I signaling, antigen presentation, and production of CTL-recruiting chemokines, suggesting that the mechanism identified in mice is similar in humans. Similar results were found in datasets for locally invasive metastatic TNBC treated with chemo and or immunotherapy, where PD-1hi macrophages correlated positively with patient responses to therapy, and for metastatic melanoma treated with nivolumab, where macrophages with high IFN-I signaling correlated with markers of CTL infiltration (CD8B) and cytotoxic activity. These results were validated across all tumor types in TCGA.
Further, enhanced expression of the tPD-L1 loss signature, characterized by repression of myeloid SHP2 activity, was observed exclusively in responders, suggesting that tumors infiltrated by PD-1/SHP2-inhibited myeloid cell progenitors with the capacity for increased IFN signaling could play a role in responses to PD-1/PD-L1 checkpoint blockade. In patients harboring these progenitors, a survival benefit was observed at late, but not initial time points, suggesting that the impact of PD-1 blockade on this patient population may limit metastatic outgrowth and support durable responses to immunotherapy.
Overall, these results support a mechanism by which tPD-L1 engages mPD-1 at sites of metastasis, activating SHP2 to antagonize the IFN-I-STAT1-CXCL9 pathway, limiting CTL tumor recruitment and supporting the development of metastases. Release of this mechanism though PD-1/PD-L1 axis blockade may therefore contribute to the long-term efficacy of treatment through the prevention of metastatic outgrowth.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, lead author Kebin Liu answered our questions.

What was the most surprising finding of this study for you?
Tumor-expressed PD-L1 (tPD-L1) did not inhibit cytotoxic T lymphocytes’ (CTLs) effector functions when only tumor cells and T cells were present in the microenvironment. Furthermore, tPD-L1 had no significant role in promoting primary tumor immune evasion. Instead, tPD-L1 activated the myeloid PD-1 (mPD-1) repressive pathway to suppress type I interferon (IFN-I) signaling and CTL tumor recruitment in lung metastases.
What is the outlook?
One of the most interesting findings is that IFN-I plays a key role in CTL recruitment to the tumor site in lung metastases. IFNɑ is an FDA-approved agent. We plan to develop a nanoparticle-based DNA therapy to force the tumor cells to express IFNa, which may be an effective therapy for patients with lung metastases.
What was the coolest thing you’ve learned (about) recently outside of work?
John is in Spain for a vacation — the trip was delayed for more than three years due to COVID! Dr. Priscilla Redd gave birth to Chloe in February, a cute next-generation scientist. Priscilla is enjoying her scientist motherhood. As for Kebin, he injured his right leg during a soccer game and has to wait for some time before he can play again.



