The role of the tumor immune microenvironment (TME) in tumor progression is increasingly appreciated, but for certain cancers, our understanding remains incomplete. Recently, single-cell analysis techniques have enabled comprehensive analysis and supported our knowledge of tumor development and treatment response. Now published in Cancer Cell, Braun and Street et al. used single-cell RNA and TCR sequencing to profile the clear cell renal cell carcinoma (ccRCC) TME along multiple disease stages.
To begin, the researchers gathered samples of ccRCC tumors and healthy adjacent tissue from 13 patients ranging from early to advanced/metastatic ccRCC. The isolated cells, of which ~95% were immune cells due to the dissociation protocol used, were then analyzed through scRNA and TCR sequencing. 39 cell clusters were identified within the TME, from lymphoid and myeloid immune populations to tumor cells and healthy kidney cells.
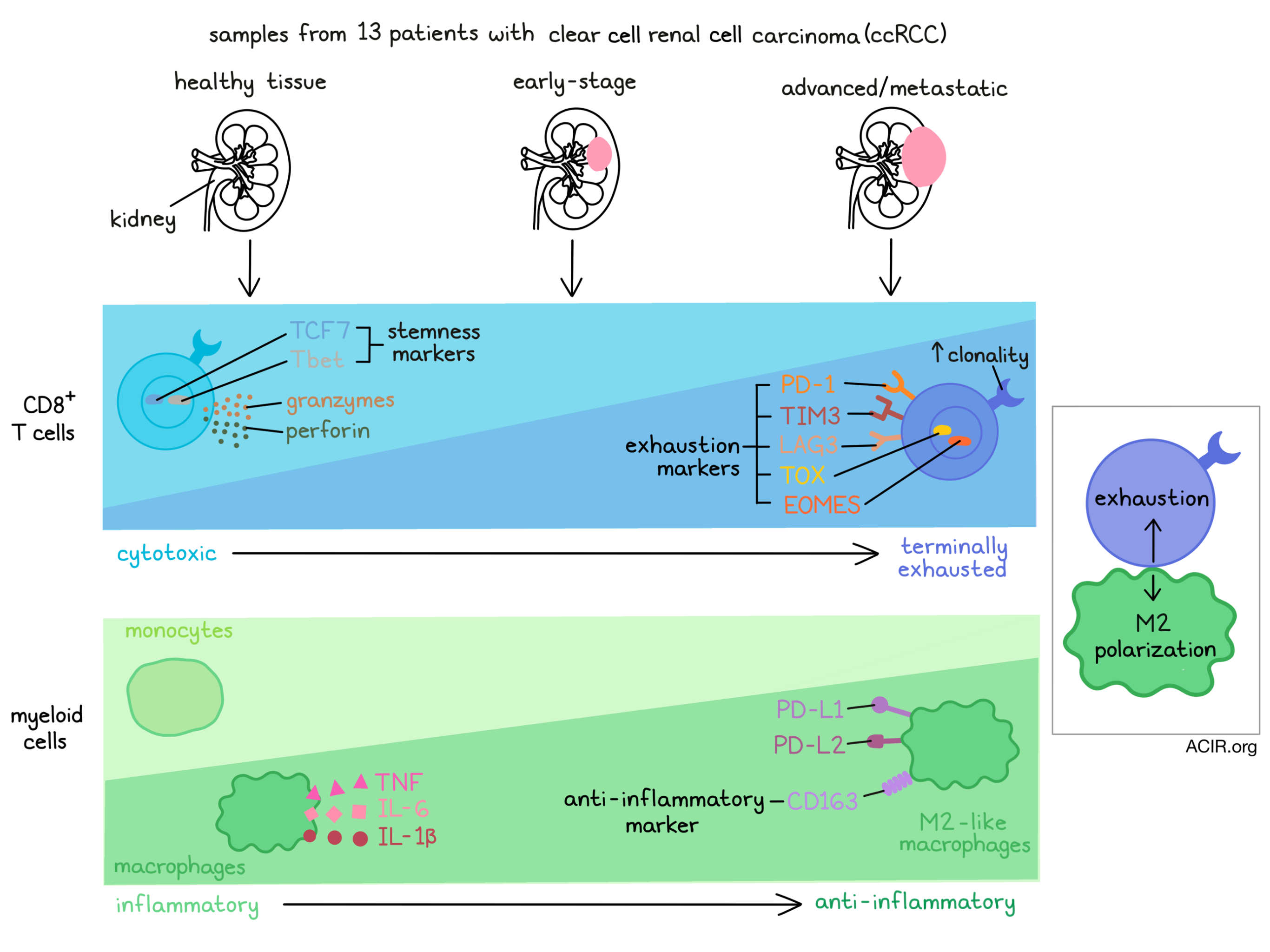
The authors first examined the tumor T cell compartment, which comprised 19 subpopulations. CD8+ T cells displayed a range of phenotypes, including terminally exhausted (high expression of PDCD1, TOX, HAVCR2), exhausted (lower expression of PDCD1 and TOX), cytotoxic/effector (PRF1, GZMH, KLRG1) and tissue-resident memory (ITGAE, ZNF683). CD4+ T cells were also distributed into several clusters, such as activated (CD69, CD40LG), central memory (IL7R, CCR7) and Treg (FOXP3, IL2RA) populations. Interestingly, the relative abundance of these phenotypes corresponded with disease stage. Particularly, cytotoxic CD8+ T cells were enriched in healthy tissue and early ccRCC, but were displaced by terminally exhausted CD8+ T cells, which were enriched in advanced and metastatic disease. More broadly, T cells displaying features of exhaustion were also enriched in later-stage ccRCC.
To map the relationships between these T cell states, the authors constructed a pseudotime trajectory. Strikingly, the position of CD8+ T cells along pseudotime matched the disease state from which the cells were isolated; CD8+ T cells from advanced/metastatic tumors (which were enriched in the terminally exhausted phenotype) were concentrated farther along the trajectory. In agreement with this result, expression of inhibitory markers (PDCD1, HAVCR2, LAG3) and ENTPD1 (encoding CD39) increased along pseudotime. Similarly, TCF7 and TBX21 (T-bet), associated with a stem/progenitor phenotype, declined along pseudotime, while the exhaustion-associated transcription factors TOX and EOMES increased. Furthermore, gene signatures of terminal differentiation, exhaustion and dysfunction all increased along the trajectory. Taken together, these results indicate progressive exhaustion and dysfunction of CD8+ T cells within ccRCC tumors.
Next, the researchers investigated TCR clonotypes associated with the observed transcriptional states. Specific clonotypes were found either within one phenotypic cluster (particular Treg clonotypes, for example) or shared among multiple clusters. Compared to healthy adjacent tissue, T cells within ccRCC tumors were highly clonal at all stages of disease, especially in metastatic tumors. Among all T cell subsets, terminally exhausted CD8+ T cells had the lowest clonotypic TCR diversity, which declined along the pseudotime trajectory. While the tumor specificity of these clonotypes could not be ascertained, a low proportion (<0.16%) were inferred to be virus-specific, based on comparisons to curated databases. With this result, along with the high expression of PDCD1 and ENTPD1, the authors speculated these cells were likely not bystanders, and may have been involved in the tumor response.
The team next considered the TME myeloid compartment. 16 clusters were identified, including monocytes, macrophages, DCs and mast cells, which each comprised additional subpopulations. Prominently, two tumor-associated macrophage (TAM) clusters were enriched in metastases. A pseudotime analysis suggested a trajectory of classical monocytes (CD14+) differentiating into non-classical monocytes (CD14-) or macrophages. Supporting this analysis, macrophage abundance increased in advanced disease, while monocytes declined. Additionally, macrophages from advanced tumors were observed later in the pseudotime trajectory and showed reduced expression of inflammatory markers (IL1B, TNF) and increased expression of anti-inflammatory markers (CD163). These results suggest that the ccRCC TME remodels towards an anti-inflammatory myeloid phenotype with progressive disease.
The researchers next studied how the observed changes in T cell and myeloid phenotypes might be linked by using CellPhoneDB, a published algorithm used to identify potential connections via receptor–ligand interactions. Compared to other T cell clusters, the terminally exhausted CD8+ T cell subset was predicted to interact highly with monocytes and macrophages. These predicted interactions included receptor–ligand pairs associated with both inhibitory checkpoints and M2-polarizing signaling (e.g., MIF, CSF1). Supporting these predictions, flow cytometry confirmed surface expression of inhibitory proteins (PD-1, TIM3, TIGIT) on ccRCC-derived T cells and their ligands on myeloid cells, which were significantly elevated compared to cells derived from healthy donors, and the expression of these ligands and receptors increased from early stage to advanced/metastatic ccRCC. Furthermore, M2-like macrophages and terminally exhausted CD8+ T cells were found in higher density in tissue sections from advanced disease, and tended to co-localize. Overall, these findings suggest that CD8+ T cell and myeloid interactions may cooperatively induce terminally exhausted T cell and M2-type macrophage phenotypes in later-stage tumors, creating an immune dysfunction circuit.
Finally, the authors tested their observations in independent ccRCC cohorts. In one cohort, terminally exhausted CD8+ T cells and M2 TAMs (assessed through mass cytometry) increased with disease progression. In another, a gene signature constructed by the researchers to encompass TAMs/terminal exhaustion coincided with advanced disease and correlated with worse overall survival. However, this signature did not predict response to anti-PD-1 therapy in ccRCC.
In summary, Braun and Street et al. comprehensively profiled the T cell and myeloid compartments of ccRCC tumors, uncovering a progressive enrichment of terminally differentiated CD8+ T cells and M2-like macrophages in later stages of disease. These findings shed light on how alterations in the makeup of the TME correlate with tumor progression, and further investigating potential causal relationships would deepen our understanding of tumor development. Additionally, studying the immune cell interactions within ccRCC and other cancers may enable prediction of therapeutic responses to immunotherapies.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, first co-author David Braun answered our questions.

What prompted you to tackle this research question?
Kidney cancer is a disease that affects 75,000 people a year in the U.S., and unfortunately, it is still responsible for about 15,000 deaths annually. While anti-PD-1 therapy forms a backbone of treatment for advanced disease, the majority of patients do not receive durable clinical benefit from immunotherapy, and the determinants of effective antitumor immunity in this disease are still largely unknown. Importantly, kidney cancer differs from other “typical” immunotherapy-responsive tumors in many important ways – it has a very modest mutation (and neoantigen) burden; generally, a high mutation burden or a high degree of T cell infiltration are not associated with improved response to anti-PD-1 therapy. Further, while foundational work has given us information about tumor-intrinsic changes that occur with cancer progression, relatively little is known about how the immune microenvironment might change with advancing disease. With these questions in mind, we used single-cell transcriptomics to try to dissect and better understand the immune microenvironment in kidney cancer and how it might change across different stages of disease – early, locally advanced, and metastatic disease.
What was the most surprising finding of this study for you?
We found that multiple arms (i.e., T and myeloid cells) of the infiltrating immune system appear to become more dysfunctional with advancing disease stage, and that, surprisingly, these arms appear to be communicating with each other. Advanced disease-stage tumors were enriched for terminally exhausted CD8+ T cells and “M2-like” macrophages, and these two immune populations interacted with one another – myeloid cells produced ligands to support T cell exhaustion, and these exhausted T cells appeared to produce factors to support an “M2-like” polarization, forming a sort of bi-directional “circuit” that supports immune dysfunction. Our hope now is that we can perturb these interactions (i.e., break the circuit) to see if we can restore effective antitumor immunity, and pave the way for new therapeutic approaches for this disease.
What was the coolest thing you’ve learned (about) recently outside of work?
With two young children at home, most of my life outside of work revolves around my daughters. While it has been an incredibly challenging year for so many people, it has also been remarkable to see how some (particularly younger) children have adapted to the “new normal.” I remember a number of months into the pandemic, my oldest daughter was doing “Zoom school” like so many, and there was a day off. She asked me why she wasn’t going to see her friends that day, and I had sort of prepared myself for a long conversation about what “the germs” meant, and that we may not be able to see friends or other family members in person for quite some time. As the conversation began, she quickly stopped me and said, “no, I mean why can’t I see my friends on the computer today?” To her, at the age of 4, that was just the “new normal”, and she adapted to it. That’s not to say we haven’t had our challenges along the way (and that I know other families have experienced many more challenges), but it was still amazing to me to see how she and my younger daughter adapted to this new normal, and how these changes have just become part of their lives



