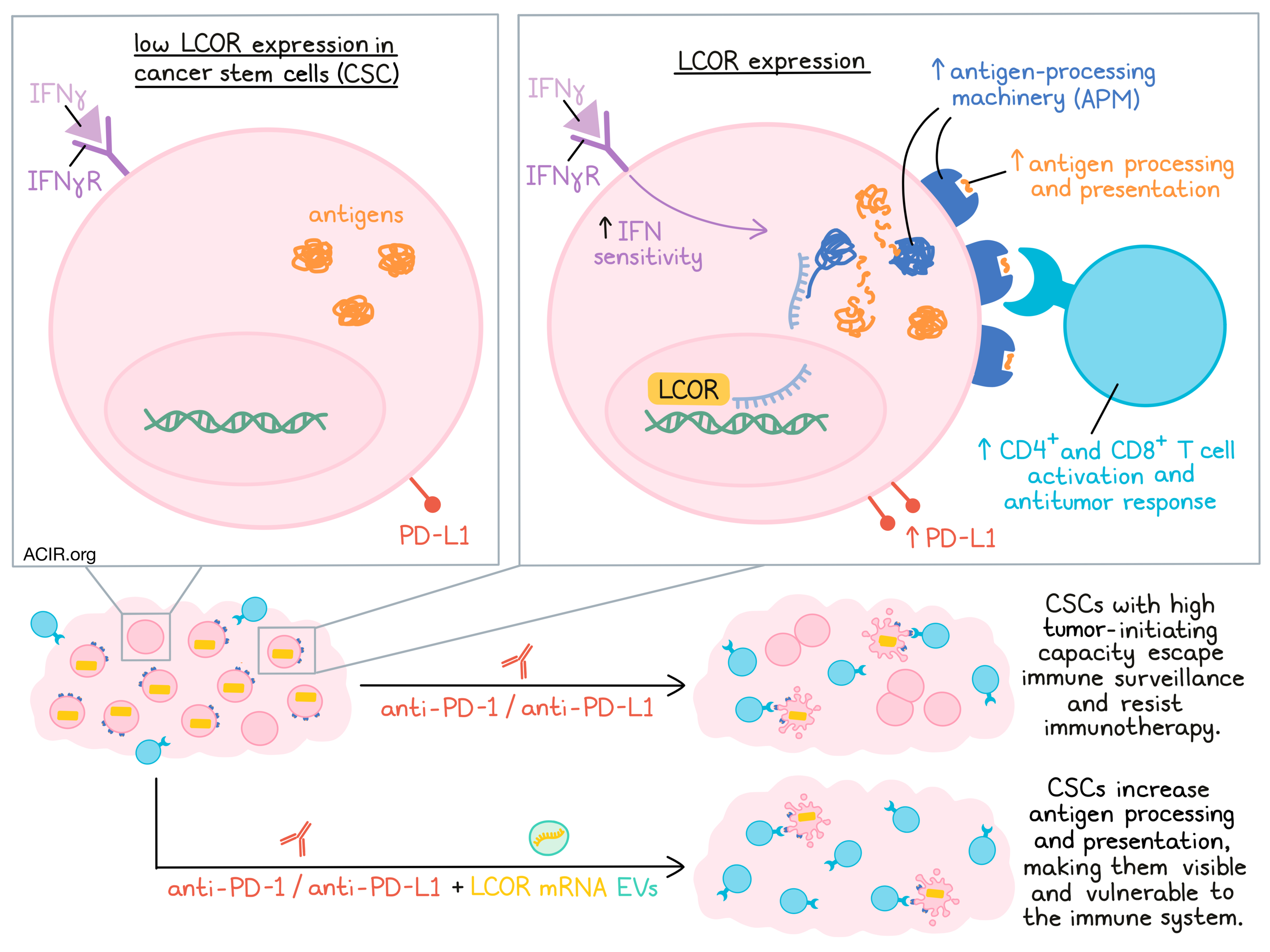
Triple-negative breast cancer (TNBC) is notoriously difficult to treat, and while anti-PD-1 and anti-PD-L1 immunotherapies are approved as treatment options, they are not particularly effective in this setting. Investigating immunotherapy resistance in TNBC, Pérez-Núñez et al. recently uncovered a novel tumor escape mechanism involving LCORlow cancer stem cells (CSCs) – and came up with a potential strategy to overcome it. Their results were recently published in Nature Cancer.
To begin, Pérez-Núñez et al. developed a mouse model of immune checkpoint blockade (ICB)-resistant breast cancer, which showed reduced antigen processing machinery (APM), IFN signaling, and mammary differentiation factors, including ligand-dependent co-repressor (LCOR). These tumor cells also maintained high PD-L1 expression, and were enriched for stem-like and tumor-initiating qualities and cancer stem cells (CSCs). In coculture with T cells, CSCs were more resistant to immune-mediated cell killing than non-CSCs, and induced less T cell activity. Similar results were observed in a humanized mouse model.
An immune-resistance signature defined in these mice was also identified in non-responders to ICB, along with an increased stem-like signature. Clinical data also showed that LCOR expression decreased, and CSC signatures increased on treatment in non-responders to ICB.
Looking more closely at CSCs, the researchers found that CSCs isolated from both patients and mice showed downregulation of APM and low LCOR expression compared to non-CSCs, and that APM gene expression progressively decreased with decreasing expression of LCOR. Suspecting that LCOR might regulate the expression of APM, Pérez-Núñez et al. employed gain- and loss-of-function mutations to show that ectopic LCOR expression induced APM pathway genes, while LCOR knockdown reduced them. Functionally, LCORlow CSCs with low APM gene expression demonstrated impaired antigen processing and presentation. They also showed increased sphere formation, and increased tumor-initiating capacity in immunodeficient mice, reflecting their inherent stem-like properties.
Next investigating the relationship between LCOR and IFN stimulation, the researchers showed that the the effects of LCOR on APM were independent of IFN stimulation, as IFN blockade did not affect the ability of LCOR to induce APM components and increase antigen presentation in vitro. Interestingly, treatment with IFNγ did increase the effects of LCOR on APM, but this effect was abrogated in LCOR-knockdown cells, suggesting that LCOR expression plays a dominant role in inducing APM, and primes sensitivity to IFN.
To determine exactly how LCOR impacts expression of APM, the team determined that LCOR binding to DNA, not to nuclear factors, was essential to the induction of APM. CHIPseq identified LCOR binding regions on the genomic region containing the MHC-I cluster and all APM genes, except for β2m. This same region is shut down in fetal mammary stem cells, suggesting a conserved gene regulatory mechanism. Additional conservation analysis revealed that LCOR is, in fact, highly conserved across vertebrates, suggesting that LCOR is likely a highly conserved transcriptional activator.
Following up on this hypothesis, Pérez-Núñez et al. identified binding motifs, and found that IFN-stimulated response elements (ISREs) were highly enriched for the top-ranked prediction motifs, particularly the IRF1 binding site. Using an ISRE reporter, the team demonstrated that LCOR could bind to ISREs and activate transcription of APM genes. The addition of IFNγ increased ISREs in LCOR+, but not LCOR- cells, while IFN inhibitors decreased ISRE activity in LCOR+, but not LCOR- cells, validating the dominant role of LCOR over IFN signaling. Similar results were observed in LCOR-overexpressing and LCOR-knockdown cells.
Next, the researchers investigated the effects of LCOR-mediated APM induction on antitumor immunity, and found that in coculture with CD8+ OT-I T cells, LCOR-overexpressing OVA+ tumor cells induced more T cell activation, and rendered tumor cells more vulnerable to T cell-mediated killing. In immunocompetent mice, LCOR-overexpressing cells grew less quickly than controls (on top of their already reduced growth due to reduced CSC-like tumor-initiating capacities), reflecting increased immunogenicity, and subsequently, increased antitumor CD4+ and CD8+ T cell infiltration and antitumor activity. In patient-derived TNBC samples, LCOR levels were highest in tumors with highly cytotoxic immune landscapes that were enriched for CD4+, CD8+, and γδ T cell signatures.
Looking at clinical data for samples of TNBC before and after treatment with anti-PD-1 or anti-PD-L1, the researchers found that LCOR expression was consistently lower in residual disease, consistent with immunoediting of cells with high antigen presentation, and that pretreatment levels of LCOR expression were associated with response to therapy. Similar results were observed in data for melanoma, which also showed an inverse relationship between LCOR expression and a resistance signature.
Exploring whether LCOR expression affects response to immunotherapy, the researchers found that control tumors were resistant to anti-PD-1, and LCOR-knockdown tumors were even more resistant. LCOR-overexpressing tumors, though, regressed in response to PD-L1, yielding complete responses in 49 out of 50 mice across several experiments. This effect was dependent on CD4+ and CD8+ T cells, and was durable in mice for up to a year of follow up. Of note, the effect of LCOR overexpression was stronger than the effect of adding the IFN inducer Poly (I:C) to anti-PD-L1. LCOR overexpression also mediated responses to ICB in a lung metastasis model.
Finally, in an effort to take advantage of LCOR in a therapeutic context, Pérez-Núñez et al. tested the delivery of Lcor mRNA using extracellular vesicles (EVs). In vitro, tumor cells transduced with Lcor mRNA showed a decrease in the CSC population, upregulation of APM genes, and increased vulnerability to CD8+ T cell-mediated killing. In a mouse model of lung metastasis, uptake and translation of Lcor mRNA was observed in most tumor cells within 5 days of EV administration. Further, mice serially treated with Lcor mRNA EVs and anti-PD-1 showed significantly longer survival and complete elimination of lung metastases compared to mice treated with the EV control and anti-PD-1.
Overall, these results support the role of LCOR as a transcriptional activator of APM that is independent of IFN signaling and primes IFN sensitivity. CSCs, which express low levels of LCOR and consequently downregulate antigen processing and presentation, are therefore uniquely equipped to escape immune surveillance and resist immunotherapy. This mechanism of resistance can potentially be targeted though delivery of Lcor mRNA, which can upregulate antigen presentation and make hidden CSCs visible and vulnerable to the immune system.
By Lauren Hitchings
Meet the researcher
This week, first author Iván Pérez-Núñez and senior author Toni Celià-Terrassa answered our questions.

What prompted you to tackle this research question?
Our lab is specialized in studying cancer stem cells (CSCs) and the tumor microenvironment (TME) in breast cancer and metastasis. Intriguingly, years ago we observed that breast CSCs were defective to respond to Interferon (IFN) due to low levels of LCOR, a factor that we found to be important in mammary epithelial cell differentiation. This IFN insensitivity led us to think that CSCs would have poor antigen presentation and would be poorly immunogenic, suggesting that tumor phenotypic heterogeneity and CSCs may influence immune evasion. Therefore we decided to investigate the implications of CSCs in cancer immunoediting, immune surveillance evasion, and immune checkpoint blockade (ICB) therapy resistance, and the output is this publication in Nature Cancer.
What was the most surprising finding of this study for you?
One of the most striking observations was the major accumulation of LCOR peaks in the MHC/APM genomic cluster of the Chr.6 across the whole genome by ChIP-seq. It was amazing to see that this highly evolutionarily conserved cluster can be fully orchestrated by LCOR directly. In addition, we demonstrated that different stem cell phenotypes have shut down the chromatin in this region, due to their lack of LCOR. In this regard, it was also surprising to see that LCOR can activate ISREs with or without IFN, representing a new mechanism of tumor immunity. Another breathtaking observation was the efficiency of ICB therapy in LCOR-expressing tumors, with complete responses in almost 100% of the cases. This was really exciting and engaged us to develop the LCOR mRNA therapy in combination with anti-PD-L1.
What was the coolest thing you’ve learned (about) recently outside of work?
IPN: In my case, I'm passionate about sports. Specifically, I loved swimming since I was a kid, but for many reasons, I left it for a long time. Lately, I've been back, and it was kind of frustrating to see that I wasn't as fit as I used to be. However, after a couple of weeks of hard work, I've reached the level again, so I realized that all things need to be persistent to get what you want!
TCT: When I was a kid, I played tennis at a high competition level, but at the age of 12 I burnt out and quit it for a long time. During the last two years with the pandemic, I re-started playing tennis and have been having the great feeling of loving it, as when I was a kid – a rejuvenating feeling



