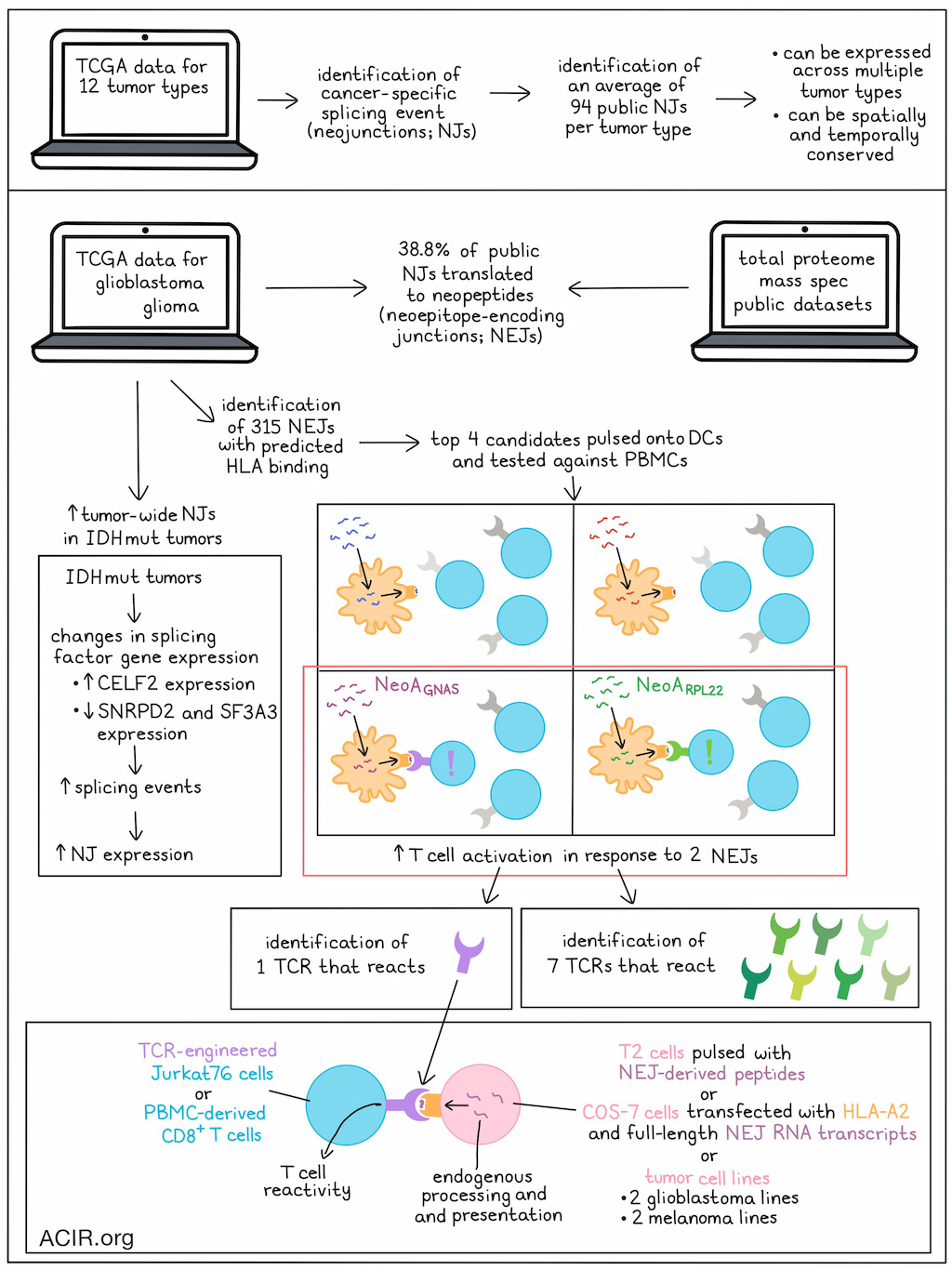
Immunotherapies targeting tumor-specific antigens (TSAs) derived from somatic mutations have limited potential in tumors with low mutational burdens. A recently discovered source of TSAs are cancer-specific splicing events (neojunctions; NJs), which have the potential to induce CD8+ T cell responses. What remains unknown is whether these NJs are conserved spatially and temporally across tumors. Kwok et al.’s recent publication in Nature examined the clonality of NJs across different cancer types to explore the existence of public tumor-wide NJ-derived TSAs as potential therapeutic targets.
To start, TCGA data were used to identify non-annotated junction reads in 12 cancer types with spatially mapped tumor samples. Then, the positive sample rate (PSR) of each junction was determined, which is the percentage of samples in each cohort expressing the NJ at a frequency of ≥1% relative to the canonical splicing junction. Public NJs were selected based on the highest PSRs in each tumor type cohort. The splicing events were considered cancer-specific if their PSR was >10% in the tumor and <1% in healthy tissue. This process retrieved an average of 94 public NJs per TCGA tumor type. NJ expression could be grouped by tumor type using unbiased hierarchical clustering, suggesting conserved patterns, and a subset of NJs was detected across multiple tumor types.
Analysis of intratumoral RNAseq data from various cancer types suggested public NJs were consistently expressed across multiple samples. Since glioma generally has high intratumoral heterogeneity, samples from intratumoral biopsies across three main glioma subtypes were assessed, demonstrating heterogeneity and emphasizing the utility of acquiring multiple biopsies. Hierarchical clustering showed that mutant isocitrate dehydrogenase subtypes (IDHmut) had significantly more tumor-wide NJs than the WT variants.
To determine whether the detected NJs were spatially and temporally conserved, metastases or recurrence samples were compared to paired primary samples. This revealed that 43.8-72.6% of NJs were present in both primary and metastasis tissues of various tumor types. Additionally, an average of 36.4% of NJs remained from primary tissue to recurrence in multiple tumor types. 79.2% of NJs in hypermutated and 82.3% of NJs in non-hypermutated gliomas were conserved in samples from recurrent tumors following temozolomide therapy. These data suggested that NJs could persist spatially and temporally.
To assess what role dysregulation of the splicing machinery plays in NJ expression, IDHmut glioma was evaluated, as IDH mutations have previously been shown to drive splicing changes. Gene set enrichment analysis of three glioma subtypes in TCGA showed that IDHmut subtypes expressed more splicing-related genes than IDHwt gliomas. Assessing splicing-related genes with a significant increase in expression in the IDHmut samples showed that CELF2 expression was correlated to NJ expression. CRISPRi and short interfering RNA (siRNA) were used to knock down CELF2 in patient-derived IDHmut cell lines. CELF2 knockdown reduced the expression of the NJ (NJacap2) that had the highest correlation with CELF2. Of the 244 NJs that were upregulated in IDHmut cases, 8.6% were downregulated in oligodendroglioma cells and 12.7% in astrocytoma cells after CELF2 knockdown. Analyzing splicing-related gene sets showing decreased gene expression in cases of IDHmut oligodendroglioma showed that downregulation of SNRPD2 or SF3A3 contributed to increased NJ expression (which was confirmed by knockdown experiments).
Analysis of public NJ expression in gliomas showed that most xenografts obtained from patients with glioblastoma (97.2%) and low-grade glioma (64.4%) expressed these public NJs. Data from glioma total proteome mass spectrometry public datasets revealed that 38.3% of public NJs were translated into neopeptides.
To determine whether the NJs could produce targetable neoantigens, NJ-derived sequences from TCGA were translated in silico to derive a NJ-based protein dataset. First, the researchers established which peptides were predicted to be presented by HLA class I. Using two prediction algorithms, neoepitope sequences were identified and ranked based on their binding potential to HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*11:01, or HLA-A*24:01. Top candidates were mapped to their original NJs, which revealed 315 neopeptide-encoding NJs (NEJs).
Kwok et al. then determined whether the detected NEJ-derived neopeptides could provoke T cell responses. Reactive CD8+ T cells populations were obtained from healthy PBMCs in a coculture assay with neopeptide-pulsed autologous monocyte-derived DCs. Four of the top NEJ candidates were assessed, and 2/4 showed neoantigen-reactivity (NeoARPL22 and NeoAGNAS). NeoARPL22 and NeoAGNAS–reactive CD8+ T cell populations were subjected to combined single-cell V(D)K and RNAseq to establish their TCR gene sequences. Seven NeoARPL22-reactive TCRs and 1 NeoAGNAS-reactive TCR were detected.
To establish whether these identified TCRs had peptide-specific reactivity, TCR-null Jurkat76 cells or PBMC-derived CD8+ T cells were transduced with the retrieved TCR α- and β-chains. The TCR-transduced Jurkat76 cells cultured with T2 cells pulsed with the neoantigen peptide showed dose-dependent reactivity, and both TCRs had nanomolar-level neoantigen recognition, showing high functional avidity. There was negligible TCR activation in the presence of high levels of control peptides. Similar data were found with the PBMC-derived CD8+ T cells, which showed T cell activation at low neoantigen-peptide concentrations. Alanine scanning mutagenesis analysis revealed no known human proteins sharing key residues required for TCR recognition, suggesting these TCRs were not reactive to normal tissue peptides.
The researchers then tested whether NEJ-derived transcripts can generate peptides functionally presented by HLA and recognized by reactive TCRs. COS-7 cells transfected with HLA-A2 and full-length mutated transcript were cocultured with TCR-transduced Jurkat76 or CD8+ T cells. Both of the TCRs induced T cell reaction against the cells expressing the cognate neoantigen, suggesting endogenous processing and presentation of the NEJ. Immunopeptidomics confirmed presentation of the neoepitopes.
Finally, Kwok et al. assessed whether tumor cells endogenously expressing NEJRPL22 and NEJGNAS could be killed by TCR-transduced CD8+ T cells. Transduced T cells were cytotoxic against GBM115 and GBM102 cells, as well as two melanoma cell lines. HLA-I blockade partially blocked the killing, suggesting the cytotoxicity was based on TCR recognition of the HLA-presented neoantigen.
The data in this study suggest NJ-derived neoantigens could serve as novel antigen targets for antitumor therapy, and given their shared nature between tumors, combinations of these antigens might be further developed as targets for off-the-shelf TCR-engineered T cell therapeutic approaches.
Write-up by Maartje Wouters, image by Lauren Hitchings



