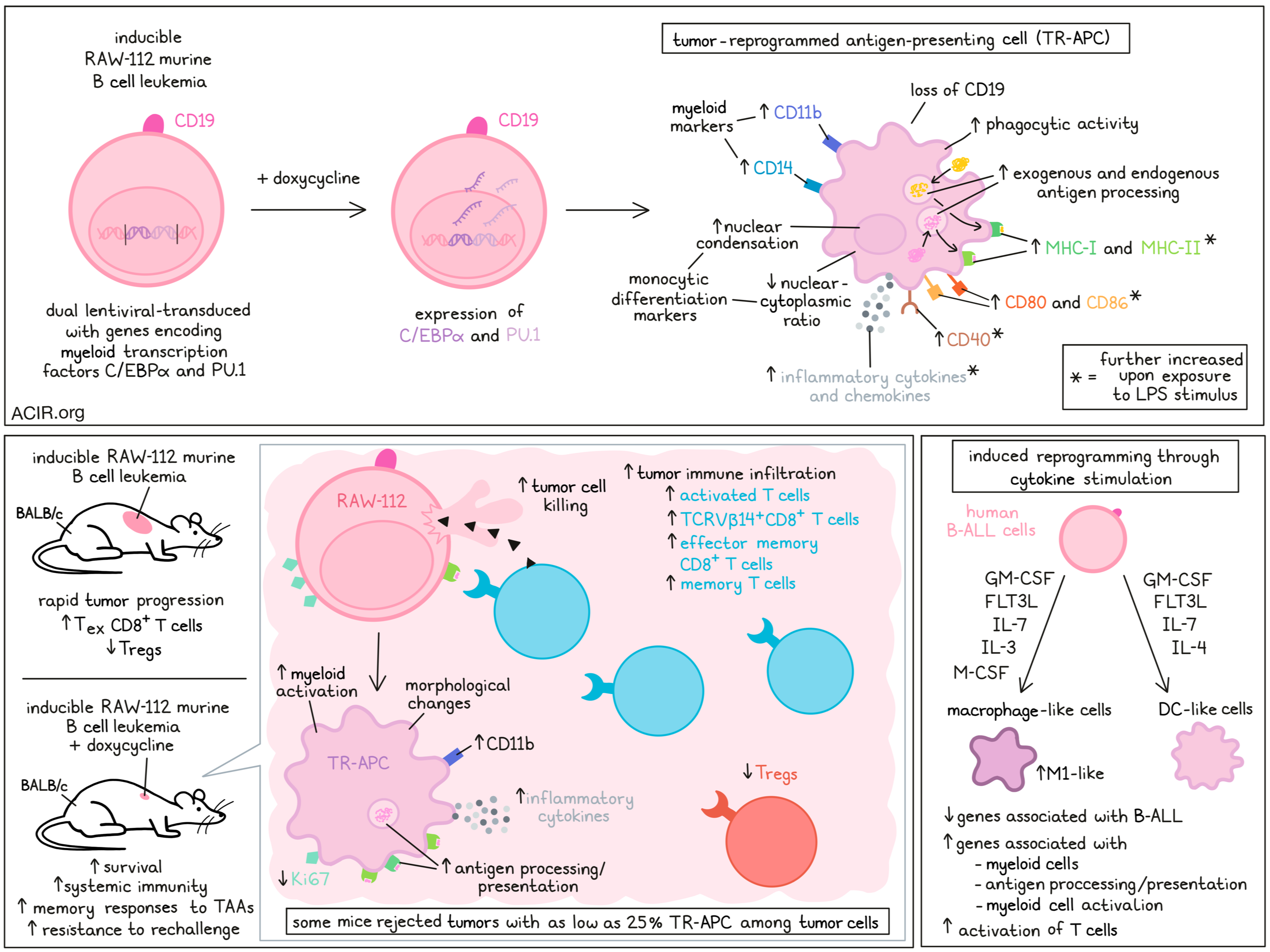
A major challenge for cancer vaccination is the choice of which tumor-associated antigens (TAAs) to target to elicit tumor-specific T cell responses, given the large tumor heterogeneity. To overcome this issue, Linde et al. developed a cancer vaccination approach in which cancer cells are reprogrammed into myeloid lineage cells, which are phagocytic and possess other APC-like properties. Their research into the behavior and therapeutic potential of such tumor reprogrammed-antigen presenting cells (TR-APCs) was recently published in Cancer Discovery.
The researchers created a doxycycline-inducible system for the upregulation of two master myeloid transcription factors (C/EBPα and PU.1) via dual lentiviral transduction into the mouse B-cell leukemia cell line RAW-112. Using this method, TR-APCs were generated after five days of induced transcription factor expression. These cells upregulated several myeloid markers, such as CD11b and CD14, and lost CD19. Furthermore, these cells had nuclear condensation and a decrease in the nuclear-cytoplasmic ratio, both of which are associated with monocytic differentiation, and cells had increased phagocytic activity. MHC-II was upregulated, and MHC-I and MHC-II were further upregulated after exposure to LPS stimulus. Similarly, expression of CD40, CD80, and CD86 was upregulated upon reprogramming the cells, and further upregulated upon LPS stimulation. TR-APC produced high levels of various inflammatory cytokines (including IL-6, IL-1β, IL-12p70, IFNγ, and TNFα) and chemokines (including CXCL10) in vitro, which was further enhanced with LPS stimulus.
Coculturing TR-APCs with DQ-OVA reagent showed that these cells could process exogenous antigens. Furthermore, when the reprogrammed RAW-122 cells were transduced with ovalbumin (OVA) and cocultured with transgenic OVA-specific CD4+ T cells, the T cells proliferated, suggestive of antigen presentation by the TR-APCs. To determine whether the cells could also present endogenous TAAs, the TR-APCs were cocultured with syngeneic T cells from BALB/c mice (the RAW-112 line is derived from BALB/c). This resulted in the activation of both CD4+ and CD8+ T cells.
Testing whether in vivo reprogramming could be conducted, NSG mice were transplanted with the inducible RAW-112 cells, and once tumors were palpable, doxycycline was injected into the tumor. Tumor cells changed morphologically in response to this, showing an increase in CD11b expression and reduced expression of the proliferation marker Ki67.
To investigate the antitumor effects of this tumor cell reprogramming approach, TR-APCs were implanted in different mouse strains. In NSG mice, the uninduced tumor rapidly progressed, resulting in short survival. Induction of TR-APCs reduced tumor progression, but was still deadly, suggesting that the therapy was not effective in absence of immune cells. In BALB/c mice, induction of TR-APCs resulted in tumor cell killing and increased survival. The approach also induced immune memory responses against TAAs, as tumors did not develop upon rechallenge with the parental cell line.
Assessing the requirement of TR-APC induction efficiency for therapeutic responses, the reprogrammable RAW-112 cells were titrated to various proportions with unmodified RAW-112 cells before inoculation. Some of the mice were able to reject tumors with only 25% of cells undergoing TR-APC induction. Furthermore, when mice were inoculated with inducible cells on one side and parental cells on the other side, TR-APCs induced systemic effects, as this one-sided induction was sufficient to reject the tumors in the contralateral flank in most mice.
Immune profiling of tumoral T cells during TR-APC generation and tumor regression showed increases in activated and memory T cell populations and a reduction in Tregs. TCRVβ gene usage profiling showed oligoclonal expansion of TCRVβ14+CD8+ T cells that were enriched for antitumor activity. Single-cell RNAseq of tumors showed an increase in total immune infiltration after TR-APCs were induced. At day 5 after induction, almost all T cell phenotypes were increased, with naive CD8+ and Th1 CD4+ T cells being the most common cell types. At day 10, naive and effector memory CD8+ T cells and Th1 CD4+ T cells were the most common infiltrating cells in these tumors. On the other hand, tumors that were not induced for TR-APCs contained mostly exhausted CD8+ T cells and Tregs.
After TR-APC induction in these tumors, there was increased myeloid cell activation and inflammatory cytokine production. The TR-APCs were enriched for gene signatures related to antigen processing and presentation, myeloid activation, and cytokine production.
To determine whether TR-APCs collaborated with endogenous APCs, the researchers made use of BALB/c mice in which cDC1s could be conditionally depleted, revealing that these cells were not required for tumor regression in response to TR-APC induction. However, when antigen presentation of TR-APCs was inhibited by a TAP-1 knockout, in the presence of cDC1s some of the mice could still eradicate the tumor cells, suggesting TR-APCs may collaborate with endogenous APCs for antigen presentation and T cell activation.
To assess whether this strategy could also be applied to solid tumors, various tumor cell lines were screened for their response to myeloid reprogramming. Multiple murine models of sarcoma and carcinoma were found to be reprogrammable. In vivo induction of TR-APCs resulted in improved outcomes in three solid tumor models, and even intrafemoral tumors could be eradicated using this therapy.
To study the applicability to patients, the researchers used human B-ALL cells to generate TR-APCs not by gene modification but by cytokine stimulation. Stimulating B-ALL cells with GM-CSF, M-CSF, IL-3, IL-7, and FLT3L created macrophage-like cells, and with GM-CSF, IL-4, IL-7, and FLT3L created DC-like cells. scRNAseq of these TR-APCs showed increased expression of myeloid genes, genes related to antigen processing and presentation, and myeloid cell activation genes, while B-ALL genes were downregulated. Most of the macrophage-characterized TR-APC cells had features of inflammatory M1-like macrophages. TR-APCs induced more activation of unmanipulated autologous T cells than original B-ALL blasts in coculture experiments, and this activation was inhibited in the presence of MHC-blocking antibodies.
These data reveal a novel approach to cancer vaccination with human translational potential that does not require identification of a predefined selection of TAAs, as it inherently contains all cell-derived antigens, including both neoantigens and self-derived antigens. It will be of interest to determine how this approach could be applied in the human setting (direct in situ reprogramming, ex vivo reprogramming, etc.) as an approach to stimulate the antitumor immune response.
Written by Maartje Wouters, image by Lauren Hitchings



