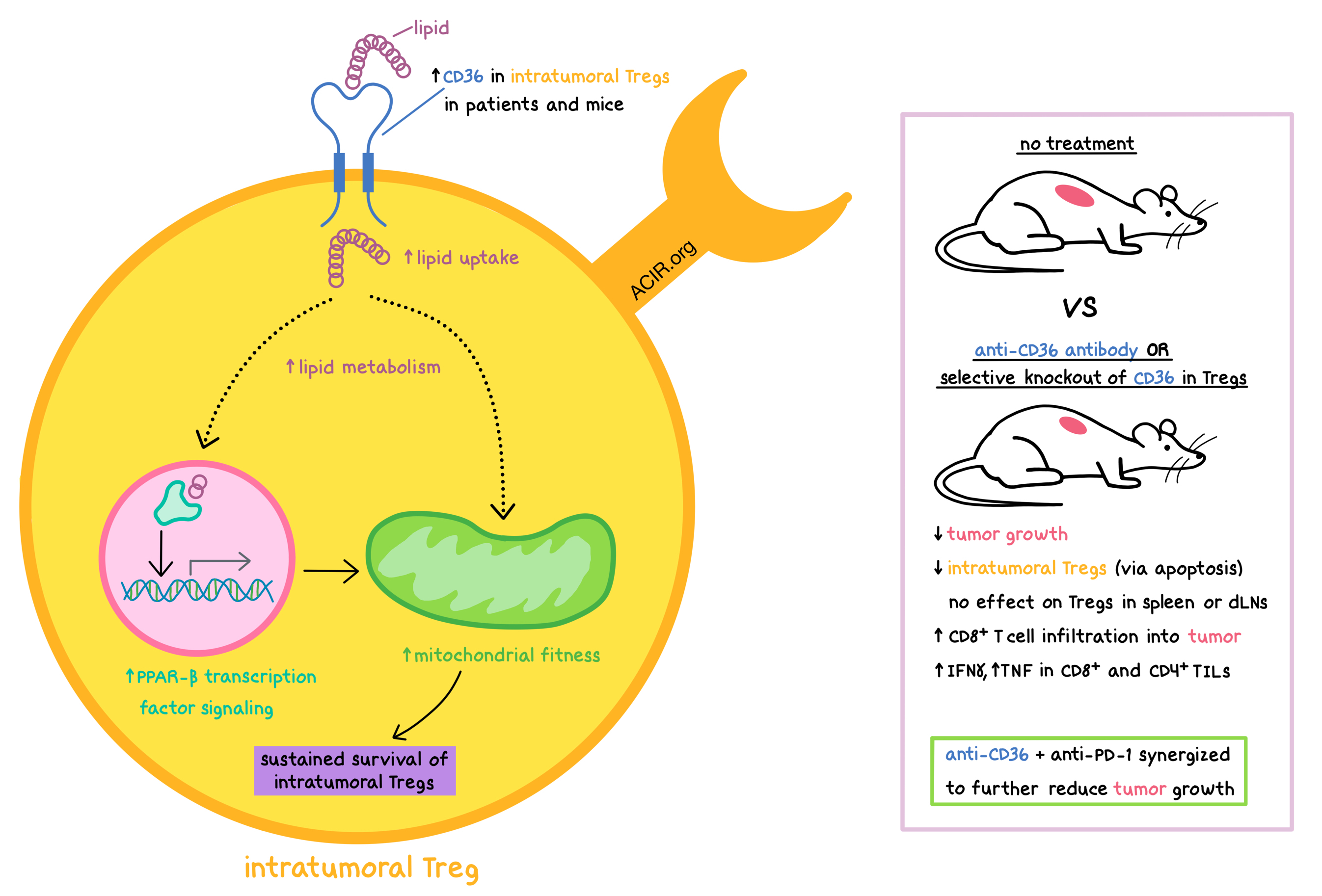
With the goal of selectively disrupting intratumoral Tregs without inducing systemic autoimmunity, Wang et al. hypothesized that intratumoral Tregs must metabolically adapt to the harsh conditions of the tumor microenvironment (TME), and therefore explored metabolic pathways that could potentially target Tregs exclusively within the TME to find actionable targets. The results, recently published in Nature Immunology, identified CD36 in intratumoral Tregs as the target of choice.
The researchers began by using RNAseq to analyze the metabolic pathways utilized by intratumoral and circulating Tregs in patients with breast cancer, and found that intratumoral Tregs upregulated genes related to lipid metabolism when compared with circulating Tregs. In particular, CD36 was upregulated in intratumoral Tregs compared to circulating Tregs in patients with breast cancer, melanoma, and non-small-cell lung cancer (NSCLC). CD36 is a scavenger receptor that plays a role in the uptake of long-chain fatty acids and oxidized low-density lipoproteins. Turning to mouse models, Wang et al. observed that intratumoral Tregs were more capable of taking up fatty acids and had higher lipid content than Tregs from other tissues (including spleen, thymus, and draining lymph nodes) in mice with YUMM1.7 melanoma, B16 melanoma, or MC38 colon carcinoma. Similar to patient data, CD36 was selectively upregulated in intratumoral Tregs in these mouse models, as well as in mice with Braf/PTEN melanoma or K-ras/p53 NSCLC. Thus, increased lipid metabolism and lipid uptake in intratumoral Tregs appears to be a conserved pathway in human cancers and mouse tumor models, and the TME appears to stimulate CD36 expression in Tregs.
Next, the researchers created mice with a knockout of CD36 in Tregs only (TregCd36-/-) and showed that CD36 is not required for the maintenance of immune homeostasis. However, CD36 was required for the increased lipid uptake and lipid content in intratumoral Tregs in YUMM1.7 melanoma-bearing mice. Tumor growth was delayed in TregCd36-/- mice bearing YUMM1.7, B16, or MC38 tumors compared with tumor-bearing wild-type (WT) mice. In addition, Tregs in the tumor, but not in the spleen or the draining lymph nodes, were reduced in TregCd36-/- mice. At the same time, the frequency of CD8+ tumor-infiltrating lymphocytes (TILs) was increased, as was the ratio of intratumoral CD8+ T cells to Tregs. A larger percentage of CD8+ and CD4+ TILs produced IFNγ and TNF, indicating that the TME of TregCd36-/- mice was less immunosuppressive. Together, these results suggest that CD36 plays a critical role in allowing Tregs to accumulate within the TME.
Further examining the role of CD36, Wang et al. found that CD36 was more highly expressed on intratumoral effector Tregs compared with intratumoral resting Tregs in melanoma-bearing mice. In patients with melanoma, a higher portion of intratumoral GITR+CD25+ effector Tregs (the most suppressive subset) expressed CD36 compared with GITR+CD25+ effector Tregs in peripheral blood from patients with melanoma or healthy donors. CD36 expression appeared to contribute to the suppressive functions of intratumoral effector Tregs, as intratumoral Tregs from TregCd36-/- mice were less capable of suppressing CD8+ T cell proliferation. However, CD36 deficiency did not affect the suppressive capacity of splenic Tregs. Co-transferred CD36-deficient Tregs were as capable as WT Tregs of suppressing naive T cell transfer-induced colitis in Rag1-/- mice (an example of peripheral inflammation). Additionally, CD36-deficient intratumoral Tregs had a slightly increased production of IFNγ and TNF. Together, these results show that CD36 specifically supports the suppressive function of Tregs within tumors.
Diving into the mechanism behind the effect of CD36 on intratumoral Tregs, the researchers saw that CD36 deficiency did not affect their proliferation, but did increase intratumoral Treg apoptosis, impair mitochondrial fitness (as indicated by reduced mitochondrial membrane potential and fewer mitochondria), and decrease oxygen consumption rates while increasing glycolytic rates, suggesting the skewing of intratumoral Treg metabolism away from oxidative phosphorylation (OXPHOS) and toward aerobic glycolysis. CD36-deficient Tregs had a decreased NAD-to-NADH ratio, which indicates impaired lactic acid metabolism, and were less viable when exposed to increased concentrations of lactic acid, which is commonly found within the TME. Furthermore, WT intratumoral Tregs had increased PPAR-β transcription factor signaling, which regulates mitochondrial activity and biogenesis, and knockout of PPAR-β in Tregs led to effects similar to CD36 knockout. Mechanistically, CD36-mediated lipid uptake activated PPAR-β pathways, which in turn supported mitochondrial fitness in intratumoral Tregs. Moreover, activation of PPAR-β pathways may also increase CD36 function. Thus, the CD36-PPAR-β signaling axis regulates metabolic programs that allow Tregs to persist within the metabolically stressful TME.
To specifically target intratumoral Tregs, Wang et al. treated YUMM1.7 melanoma-bearing mice with an anti-CD36 antibody. The treatment prevented CD36-mediated uptake of fatty acids and lipoproteins, reduced tumor growth, reduced intratumoral Tregs (by promoting their apoptosis) without affecting Tregs in the spleen and draining lymph nodes, increased CD8+ T cell infiltration into the tumor, and increased the production of IFNγ and TNF by CD8+ and CD4+ TILs. The antitumor effect was dependent on targeting CD36 on Tregs, and not on other CD36-expressing cells. Anti-CD36 synergized with anti-PD-1 to further restrict tumor growth in mice with Braf/PTEN or YUMM1.7 melanomas.
In this study, Wang et al. demonstrated that targeting CD36 in Tregs selectively affects metabolic processes in intratumoral Tregs without causing systemic autoimmunity, rewires the TME toward less suppressive conditions, and allows PD-1 blockade to reinvigorate intratumoral CD8+ and CD4+ T cells for an antitumor response. While the exact mechanism linking metabolic adaptation and suppressive activity in intratumoral Tregs remains to be elucidated, the results of this study support the development of therapies targeting CD36 for the treatment of human cancer.
by Anna Scherer
MEET THE RESEARCHER
This week, lead author Ping-Chih Ho and first author Haiping Wang answered our questions.

What prompted you to tackle this research question?
PCH: We know that targeting intratumoral Tregs is important for cancer immunotherapy; however, this is an unmet need in most of current trials. This prompted us to tackle this challenge.
HW: Having a different background from my master study, I began to learn immunology four years ago. Over the past decades, the understanding of immunosuppressive role of Treg cells in the tumor microenvironment has rapidly progressed. Therefore, in the beginning, we wanted to focus on Treg cell targeting in the tumor microenvironment in order to improve the antitumor immunotherapy. Overtime, we realised that the benefits of the potential approaches are largely impeded by systemic Treg cell targeting, resulting in the induction of autoimmune responses. Meanwhile, recent studies on Treg cell metabolic reprogramming suggest an assumption of tissue-resident Treg metabolic adaptation. All that previous research encouraged us to start to discriminate intratumoral Treg cells from those in other tissues. Thus, based on existing data, we were able to find our target, CD36, on Treg cells in the tumor microenvironment.
What was the most surprising finding of this study for you?
PCH: We were surprised when we realized that targeting CD36 could selectively demolish intramural Tregs because of high lactate utilization by intratumoral Tregs.
HW: During the study, the most striking thing for me was that intratumoral Treg cells significantly increased lipid metabolism as compared to Treg cells residing in other tissues, which perfectly matched our hypothesis about Treg cell metabolic adaptation in response to different environments. On the basis of this finding, we found that intratumoral Treg cells could be disrupted by targeting CD36. I was further surprised that the inhibition of CD36 enhanced antitumor immune response, indicating a potential target in cancer immunotherapy. Yet, we still don’t know exactly which factors in the tumor microenvironment selectively induce CD36 expression on Treg cells, and whether CD36 targeting also affects other cell metabolic reprogramming in the tumor microenvironment. Hence, a lot of work still needs to be done.
What was the coolest thing you’ve learned (about) recently outside of work?
PCH: How to make fried chicken more tasty!?
HW: I have been into drawing and painting for a long time. I love watercolour most, but recently, I started to explore more painting techniques, such as oil and mark paintings. Most of the time when I am alone, I pick up pens and papers that I could find and draw whatever I see. My friends always get me painting tools as gifts so that they can get my works in return. I won some awards from drawing and painting, but the most beautiful thing that I gain from painting is the peace of my mind.



