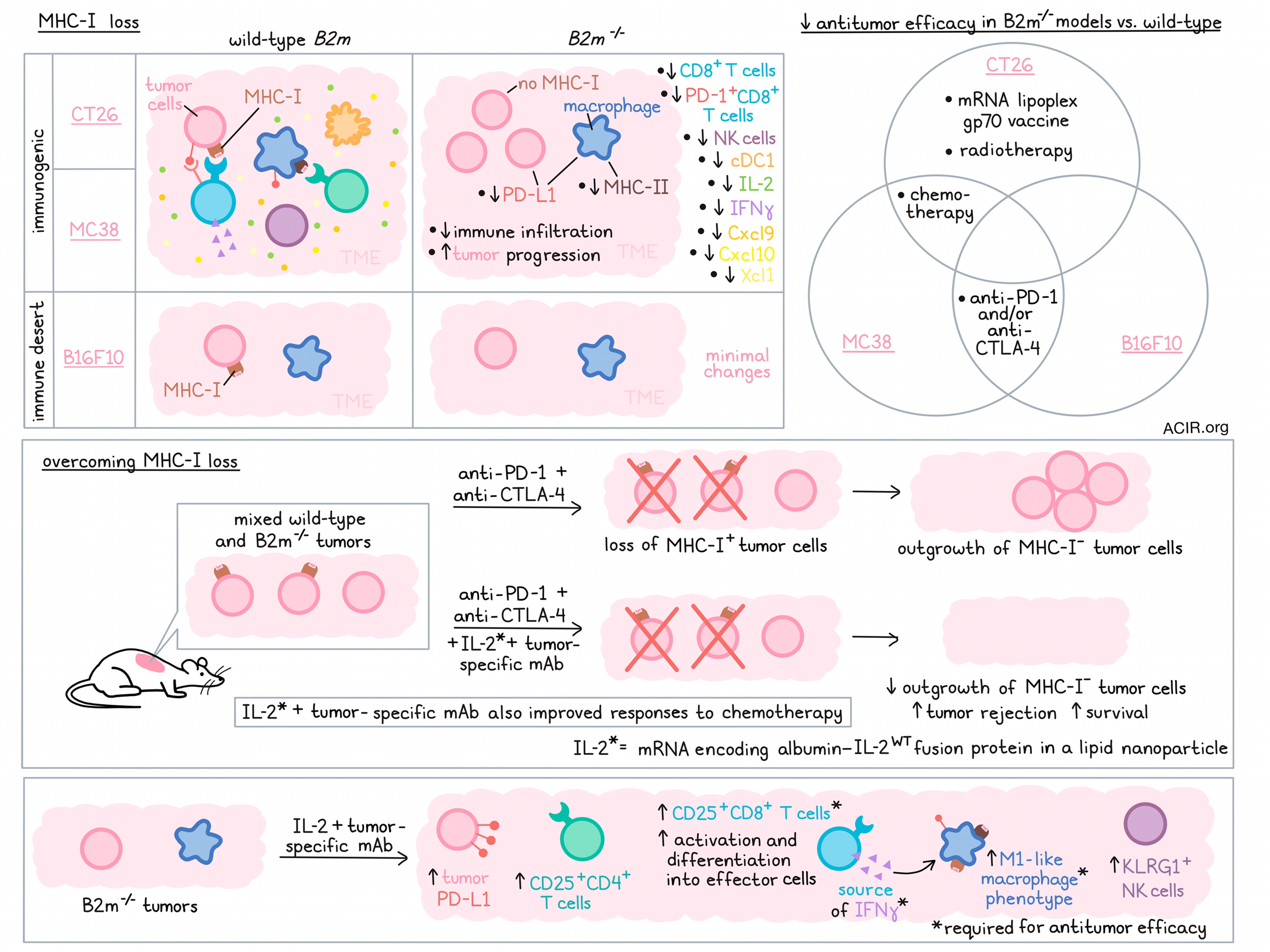
Loss of MHC class I expression on tumor cells is a frequently observed immunotherapy resistance mechanism. The increased use of immune checkpoint blockade (ICB) for the treatment of solid tumors has resulted in more therapy-resistant MHC-I-deficient tumors. To better understand the mechanisms behind these effects and determine how to overcome this response, Beck et al. investigated murine models lacking MHC-I in a recent Cancer Cell publication.
Since deletion of B2m genes results in complete loss of MHC-I surface expression, the researchers deleted the gene from three murine tumor cell lines. The experiments used two immunogenic tumors (CT26 and MC38) and one immune desert-like tumor (B16F10). Comparing the changes in the tumor microenvironment (TME) during the growth of wild-type (WT) and B2m-/- tumors, a gradual decrease in immune cell infiltration was detected in CT26- B2m-/- and MC38-B2m-/- tumors. There were decreases in CD8+ T cells, NK cells, and conventional type I dendritic cells (cDC1s). The B16F10 model showed little difference, as the WT variant already had little immune infiltration. CT26-B2m-/- and MC38-B2m-/- tumors progressed faster than their WT versions. Both models had a significant loss of PD-1+CD8+ T cells compared to WT versions. Furthermore, expression of IL2 and Ifng was reduced, and tumors had reduced levels of IFNγ-induced chemokines Cxcl9, Cxcl10, and Xcl1 (involved in cDC1 recruitment). PD-L1 was downregulated on tumor cells, and PD-L1 and MHC-II expression were downregulated in tumor-infiltrating macrophages (TAMs).
To address how B2M deficiency affects therapy outcomes, the tumor models were treated with anti-PD-1 and/or anti-CTLA-4 ICB. In the WT B16F10 and MC38 models, treatment with ICB resulted in more tumor rejection and longer survival, while therapy did not affect the B2m-/- versions of these tumors. Furthermore, treatment of the WT CT26 model with an mRNA-lipoplex vaccine encoding the antigen gp70 resulted in prolonged survival and rejection of 40% of tumors, while it did not impact the B2m-/- model. Finally, the effects of platinum-based chemotherapy in the MC38 and CT26 tumors were reduced in the B2m-/- versions, and local radiotherapy did not impact survival as much in the CT26-B2m-/- models as it did for WT tumors, together suggesting broad therapy resistance in B2M-deficient tumors.
Next, the researchers assessed the impact of IL-2 treatment in these models. An mRNA coding for a long-acting albumin-fused IL-2 formulated in a lipopolyplex nanoparticle (“IL-2”) and delivered retro-orbitally was used for experiments, as it has a longer half-life and is safer than repeated high doses of IL-2 protein alone. In contrast to other recent studies, WT IL-2, with high affinity to CD125-containing IL-2 receptor complexes, was used to optimize T cell stimulation. The experiments were conducted in MHC-I-deficient tumor models for which a tumor-specific mAb treatment was available. MC38 cells were engineered to express Her2/neu (MC38-Her2-B2m-/-) and so could be treated with anti-Her2 mAbs, and Trp1+B16F10-B2m-/- cells were targetable with anti-Trp1 mAbs. Combination treatment of IL-2 and anti-Trp1 mAbs of Trp1+B16F10-B2m-/- tumors resulted in tumor rejection and prolonged survival in 60% of mice, and 50% of MC38-Her2-B2m-/--bearing mice had long-term survival after combination therapy. These effects were significantly better than monotherapy of either agent.
Given that human tumors are heterogeneous, Beck et al. investigated therapy response in models inoculated with a combination of WT and B2m-/- cells. In mice treated with anti-PD-1 and anti-CTLA-4 ICB, an increase in the proportion of MHC-I-deficient tumor cells from 25% to 90% was observed, suggesting depletion of the MHC-I+ cells. When mAb/IL-2 treatment was added to ICB, it almost entirely prevented the growth of MHC-I-deficient cells, resulting in a tumor rejection rate of 60%, and improving survival. Additionally, the mAb/IL-2 treatment also resulted in improved chemotherapy responses.
Evaluating the mechanisms behind this reversal in treatment resistance, the researchers found that treated B2m-/- tumors showed evidence of TME remodeling. Tumors treated with IL-2 alone or IL-2/mAb had higher proportions of CD25+CD4+ conventional T cells, CD25+CD8+ T cells, and KLRG1+ NK cells. Gene expression analysis showed that IL-2 and IL-2/mAb treatment upregulated Ifng, Il2, and chemokines Cxcl9, Cxcl10, Csf1, and Ccl2. Furthermore, it upregulated tumoral PD-L1 expression.
Depletion experiments were conducted to understand the importance of various immune populations in the therapy response. The efficacy of mAb/IL-2 was not impacted when CD4+ T cells or neutrophils were depleted, and depletion of NK cells had limited impact. However, CD8+ T cell depletion abolished therapy efficacy. Similarly, neutralizing IFNγ or depleting macrophages almost completely eliminated the therapeutic effects.
Zooming in on the cell types that were essential for the therapeutic effects, the molecular profiles of tumoral CD8+ T cells and TAMs were evaluated. CD8+ T cells in the IL-2- and mAb/IL-2-treated mice had upregulated levels of Tbx21, Klrg1, Il2ra, Il2rb, effector molecules, co-inhibitory and costimulatory receptors, and IFN response genes, suggestive of activation and differentiation into effector cells. Furthermore, mAb/IL-2 upregulated genes involved in TCR signaling.
Macrophages skewed to the proinflammatory M1-like phenotype after treatment as they had increased CD206, PD-L1, and MHC-II expression, and higher expression of genes related to tumor-supporting immune regulators, T cell-attracting chemokines, inflammation-associated markers, tumoricidal mediators, and antigen presentation. The costimulatory ligands CD80 and CD86 were highest expressed in the mAb/IL-2-treated group. scRNAseq analysis of various cell populations revealed that CD8+ T cells were the primary source of IFNγ in response to therapy, and CD8+ T cell depletion or IFNγ neutralization prevented TAMs from acquiring the M1-like phenotype.
In mice with ovalbumin-expressing B16F10-B2m-/- tumors, IL-2 or mAb/IL-2 treatment significantly enhanced the presentation of the MHC-I-restricted SIINFEKL antigen. Intratumoral CD8+ T cells from the MC38-Her2-B2m-/- tumor-bearing mice treated with mAb/IL-2 did not recognize MC38-Her2-B2m-/- cells in vitro, but released large amounts of IFNγ when cocultured with MC38-Her2 or Rpl18-pulsed bone marrow-derived DCs (Rpl18 is a highly expressed neoantigen in MC38 tumors), suggesting that CD8+ T cells directed against tumor neoantigens were present in the TME. Finally, when intratumoral TAMs were cocultured with Rpl18-specific CD8+ T cells from non-tumor bearing mice immunized with mRNA encoding these neoantigens, the Rpl18-specific CD8+ T cells released IFNγ, suggesting the macrophages had processed tumor-expressed Rpl18 in the tumor for cross-presentation.
Altogether, loss of MHC-I expression results in tumors with immune desert characteristics, which can be overcome by IL-2 and mAb treatment, which stimulates CD8+ T cells and macrophage polarization. If these data can be confirmed in the human setting, combination strategies that involve IL-2 and tumor antigen-specific mAb therapy might help prevent or overcome MHC-I deficiency induced by ICB treatment, allowing for a larger proportion of patients to benefit.
Write-up by Maartje Wouters, image by Lauren Hitchings



