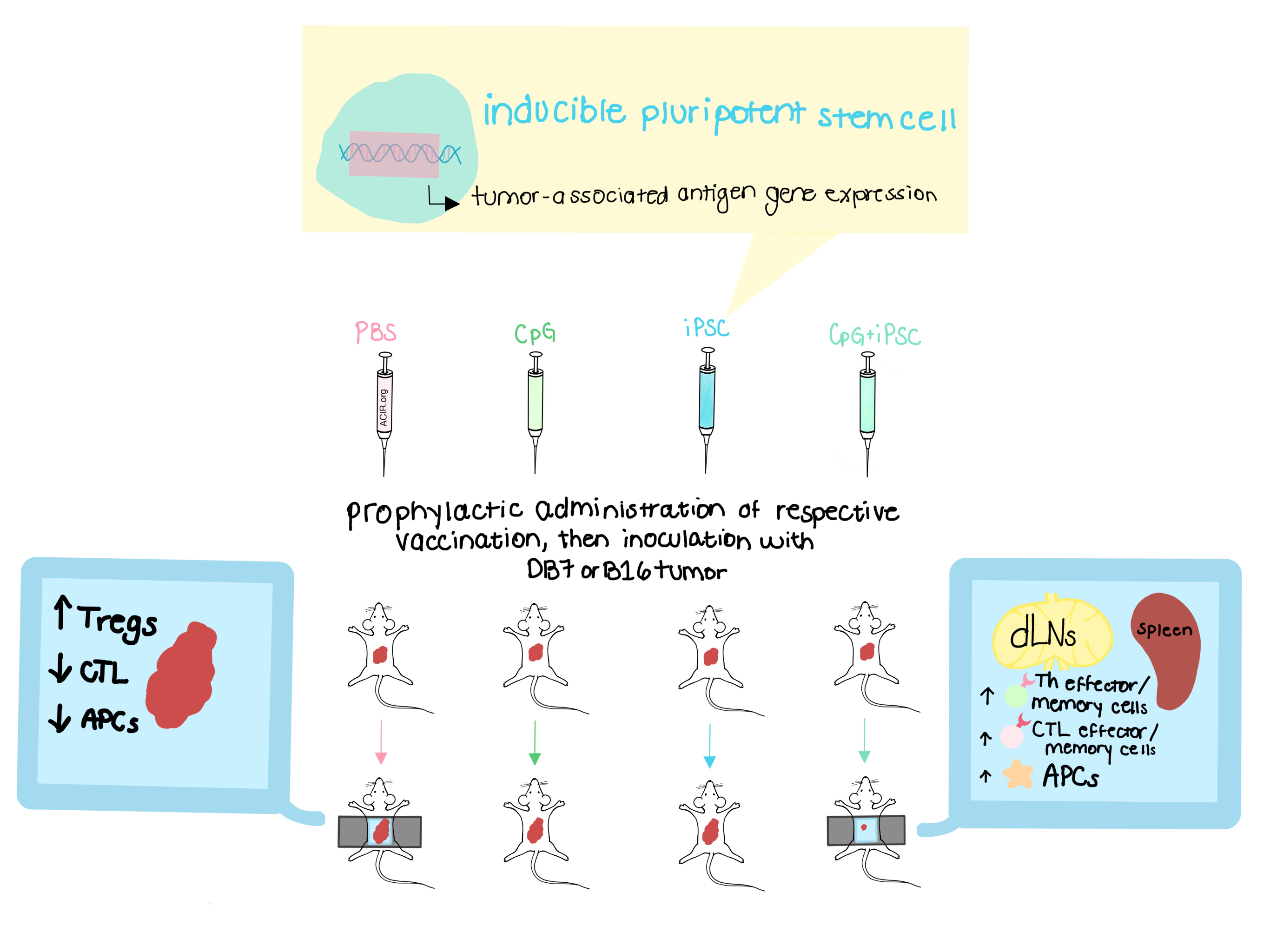
Induced pluripotent stem cells (iPSCs), lineage-specific adult cells that are reprogrammed to an earlier pluripotent state, bypass the ethical controversy associated with embryonic stem cell use. Because iPSCs and cancer cells overlap in the expression of many tumor-associated genes, iPSCs can prime the immune system for both known and potentially unknown tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). With this rationale in mind, Kooreman et al. explored the use of iPSCs as a generalizable vaccine in multiple models of cancer in their paper published in Cell Stem Cell.
Using the Toll-like receptor ligand CpG (“C”), previously shown to be an effective adjuvant in association with a whole-cell vaccine, and irradiated iPSCs from murine FVB cells (“I”), the team began their work by combining CpG with FVB strain iPSCs (C+I). After optimizing the immune response with once-weekly injections of C+I for four weeks, 40 FVB mice were vaccinated with either PBS, C, I, or C+I. After four weeks of vaccinations, all mice were subcutaneously injected with DB7 breast cancer cells. All mice initially developed tumors at one week, but tumors regressed in 7 of 10 mice in the C+I group. All other groups had progressive tumor growth.
Four weeks after DB7 tumor cell inoculation, five mice from each group were sacrificed for further study of the spleen, blood, and draining lymph nodes (dLNs); the remaining five mice from each group were maintained for long-term survival studies. Mice treated with C+I had significant increases in effector and memory cytotoxic CD8+ T cell populations in both the spleen and dLNs. Splenocytes showed an increased release of IFNγ in response to DB7 tumor lysate, demonstrating tumor specificity. Mature antigen-presenting cells (APCs) and helper T cell populations were also increased in the dLNs of C+I-treated mice. Two of the five C+I-treated FVB mice survived for one year; at that time, both mice had antibody titers against iPSCs and DB7, similar to those seen before the extended incubation period, and were able to completely reject reintroduced cancer cells.
To analyze the effect of the iPSC vaccine in other models of cancer, the same experiments were repeated in B16F0 melanoma mice with C57BL/6 iPSCs. At two weeks, tumor growth was significantly lower in the C+I mice. Flow cytometry demonstrated a significant decrease in CD4+CD25+Foxp3+ Tregs in the blood, an increase in mature APCs, and an increase in effector and memory Th cells in dLNs. In a third variation of the same experiment, performed in the AC29 mesothelioma murine model, tumor regression was successfully predicted by B and T cells expressing IL-2, IL-4, and IL-5 in C+I-vaccinated mice. Importantly, these mice had significantly lower systemic cytokine levels than PBS controls, pointing to a localized immune response and a potentially low risk of organ toxicity.
To demonstrate that the immune responses were due to antigens shared by both the cancer cell line and iPSCs – “two-way immunity” – the researchers turned to adoptive T cell transfer. T cells were prepared from C+I-vaccinated and vehicle-vaccinated mice and transferred into orthotopic DB7 tumor-bearing mice. Cancer immunity was demonstrated by a significant reduction in tumor size in the mice receiving the adoptive transfer of C+I isolated T cells compared with controls. Separately, tumor-experienced lymphocytes (TELs) were extracted from dLNs near the DB7 tumor site of C+I-vaccinated mice and vehicle-vaccinated mice and adoptively transferred into NOD-SCID mice inoculated with iPSC cells. NOD-SCID mice that received TELs from C+I-treated mice exhibited significantly reduced teratoma size, demonstrating iPSC immunity.
For their final experiment, Kooreman et al. investigated the effectiveness of the C+I vaccine as an adjuvant after tumor resection. B16F0 cells were injected into the lower backs of C57BL/6 mice and the tumors were R1-resected (microscopic residual tumor) or R2-resected (macroscopic residual tumor) two weeks later. With two adjuvant rounds of C+I, R2-resected mice had no visible tumors within their resection area, while vehicle-vaccinated mice did. After four weeks of vaccination with either C+I or CpG alone, R1-resected mice showed reduced tumor load in dLNs. Interestingly, only C+I generated significantly lower tumor recurrence in areas farther from the vaccination site, indicating the systemic effect of C+I vaccination.
Results of the experiments performed by Kooreman et al. demonstrate the effectiveness of iPSCs as a whole-cell prophylactic vaccine targeting known and unknown tumor-associated antigens in multiple models of cancer. While the results can’t be directly translated from mouse models to human studies, iPSCs hold promise, particularly as a rapidly available adjuvant therapy following primary tumor reduction.
by Brynn Vessey



