
Last week, the ACIR team attended the virtual 18th CIMT Annual Meeting. This week’s extensive special feature covers select talks from the conference. We are proud to recognize Takeda for sponsorship of our coverage for this conference.
We have organized the content by topics below.
Keynote address
Ugur Sahin
Tumor microenvironment and effective immunotherapy
Sjoerd van der Burg
Vincenzo Bronte
Ping-Chih Ho
Göran B. Jönsson
Andrea Schietinger
Darrell Irvine
T cell therapies
Tobias Feuchtinger
Johanna Olweus
Yvonne Chen
Neoantigens and cancer vaccines
Yardena Samuels
Robert Seder
Alex Rubinsteyn
Patrick Ott
Microbiome
Laurence Zitvogel
Hassane M. Zarour
Keynote address
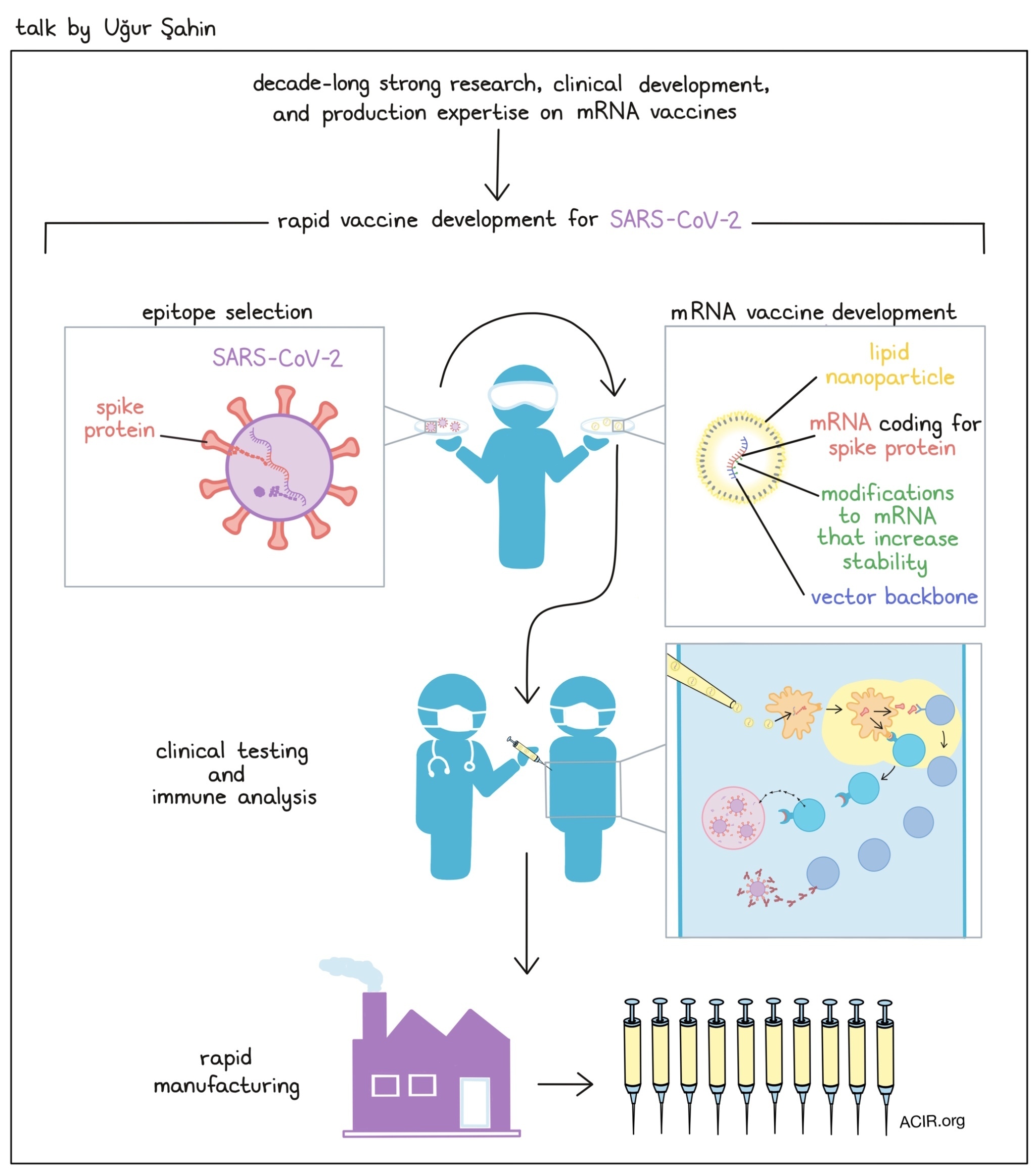
Scientific journey of developing an mRNA coronavirus vaccine- Ugur Sahin - BioNTech, Germany
With decade-long strong research and clinical development and production expertise on mRNA vaccines, Ugur Sahin and his team were able to develop, in lightspeed, a COVID-19 vaccine that is currently being administered to people in over 90 different countries of the world. Although mRNA was recognized as an immediate carrier of genetic information in 1961, it took more than 20 years before production of mRNA by in vitro transcription (IVT) became possible. Ten more years passed before Eli Gilboa, in 1996, published a study in which he used IVT RNA-pulsed dendritic cells (DCs) or, as a control, just IVT RNA, as cancer vaccines in mice – a result that inspired Sahin onto a path of dedicated and systematic research. Sahin later found that immature DCs take up mRNA in a spontaneous manner by macropinocytosis, and hypothesized that in vivo delivery of mRNA into DCs in lymphoid tissues may overcome the issue of low mRNA stability and improve translation in human dendritic cells. He evaluated multiple independent modifications of the mRNA itself that could increase the stability and translational efficacy of mRNA vaccines, and saw that by combining independent improvements and exploiting the inherent adjuvant activity of mRNA vaccines, the efficacy and potency of the vaccine was increased 5,000-fold. Intranodal injection of “naked” mRNA led to expansion of antigen-specific CD8+ and CD4+ T cells and therapeutic antitumor immunity. To facilitate i.v. delivery of the mRNA vaccines into lymphoid-resident DCs, Sahin and his team rationally designed lipo-nanoparticle-formulated mRNA with a size of 200-300 nm and a charge ratio of close to neutral. At this point, Sahin and his team had gained extensive knowledge about the impact of different mRNA modifications and had generated a variety of vector backbones that could be used for different medical applications. They have attacked cancer with mRNA vaccines targeting shared tumor-associated antigens or mutant neoantigens, and used RNA vaccines for in vivo expansion of CAR T cells. They have also moved beyond cancer by developing vaccines for prevention of infectious diseases and for the induction of antigen-specific tolerance in autoimmune disease (an application which required modification of the base composition of the mRNA to overcome its inherent adjuvanticity). The mRNA vaccines were either delivered as lipoplexes or lipo-nanoparticles, and rational design could guide the type of immune response to be elicited (e.g., cytotoxic CD8+ T cells and Th1 CD4+ T cells, expanded CAR T cells, neutralizing antibodies, or antigen-specific Tregs). The company Sahin founded and built, BioNTech SE, mastered the techniques of mRNA selection and rapid production. When the outbreak of SARS-CoV-2 struck in Wuhan, China in late 2020/early 2021, Sahin’s plans for 2021 pivoted, and he and his team saw the need to quickly develop a vaccine to keep this pandemic in check. They learned from vaccines that were developed against the prior SARS-CoV virus, which was active between 2002 and 2004, that neutralizing antibodies could be developed against the spike protein. In less than 3 months, they tested different immunogen and vector backbone combinations and used their already available technology to develop a lipid-nanoparticle vaccine against SARS-CoV-2. When injected into the muscle, this vaccine delivers the mRNA to the lymph node, where it gets translated and elicits neutralizing antibodies and T cell responses. Epitope mapping revealed HLA-specific, T cell reactivity against different regions of the spike protein, including the receptor binding domain. Up to 25% of peripheral T cells were induced by the vaccine. Phase 3 trial data suggested a rapid onset of protection against COVID-19 as early as 12 days after the first dose, and 95% of all subjects were protected against COVID-19 after having received 2 vaccine doses. The researchers continue to test the efficacy of the vaccine against virus variants, most of which are targeted as effectively as the initial strain. With their already existing expertise, Sahin and colleagues were able to scale up the production capacity by a factor of 300,000. The fastest vaccine development against a pathogen in medical history, the BioNTech SE/Pfizer vaccine against SARS-COV-2 has benefited from the versatility of mRNA and the strong expertise Sahin and his team has developed around mRNA vaccines.
Tumor microenvironment and effective immunotherapy
Chemokine-driven spatial organization of immune cell microaggregates in oropharyngeal carcinomas containing tumor-specific T cells- Sjoerd van der Burg - Oncode Institute and Leiden University Medical Center, The Netherlands
The presence of human papillomavirus 16 (HPV16) in subsets of oropharyngeal squamous cell carcinomas (OPSCC) provides a unique opportunity to study tumor-specific T cells in these cancers, and identity features of immune reactivity and the tumor microenvironment to develop new rationales for immunotherapy. To identify immune states associated with better clinical outcomes, Sjoerd van der Burg compared immune reactivity of HPV-specific CD4+ and CD8+ TILs in HPV16+ tumors and TILs in HPV16- tumors, and found that in HPV16+ OPSCC patients, in about two-thirds of patients, TILs specific for the HPV E6 and E7 oncoproteins could be detected, and the presence of HPV-specific TILs was associated with better clinical outcome. Nanostring IO360 analysis of 24 OPSCC samples revealed upregulation of genes associated with tertiary lymphoid structures (BLK, CCL12, and LTB), DC and T cell activation, and positioning in immunoreactive (IR+) patients, suggesting the presence of hot immune signature. In non-immunoreactive (IR-) patients, immunosuppressive gene signatures were upregulated. On further analysis, higher expression of lymphotoxin B (LTB), which is associated with spatial organization of cells, strongly correlated with adaptive immunity (CD4+ T cells, CD8+ T cells, and DCs) and improved survival in this patient cohort, as well as in an independent cohort of 75 HPV16+ OPSCC patients. To analyze the effects of high LTB expression on tumor microenvironments, van der Burg and colleagues used imaging mass cytometry and identified correlations between numbers of CD4+ and CD8+ T cells vs. CD14+ myeloid cells /CD11c+ DCs and spatial interactions between T cells (CD4+ and CD8+) and DCs in IR+ OPSCC. In IR- OPSCC, spatial interaction between CD8+ T cells and M2 macrophages and between CD4+ TGFβ+ T cells and TGFβ+ myeloid cells were observed. To determine the impact of these interactions on T cells, van der Burg and colleagues performed scRNAseq and found expansion of two CD8+ T cell subclusters expressing CCL4 and XCL1 (a DC attractor), and one CD4+ T cell cluster expressing LMNA (encoding lamnin, a protein involved in DC contact) in IR+ OPSCC. CCL4 levels correlated with signatures associated with adaptive immunity, CD8+ T cell levels, and improved survival of patients with HPV16+ OPSCC.
Distinct pathways in tumor-infiltrating myeloid cells for their pro-metastatic and immune suppressive activities- Vincenzo Bronte - University of Verona, Italy
Metastasis is a multi-step process that requires the involvement of several cells that are present in the tumor stroma, with tumor-associated macrophages (TAMs) being major regulators. Vincenzo Bronte and colleagues found that disabled homolog 2 mitogen-responsive phosphoprotein (DAB2) is highly expressed in TAMs. DAB2 deletion in myeloid cells had little impact on primary tumor growth, but significantly reduced metastasis formation in multiple orthotopic or autochthonous tumor models. Single cell RNAseq analysis of wild-type and DAB2 knockout (KO) tumor-infiltrating TAMs revealed upregulation of genes involved in tissue remodeling and M2 polarization in wild-type TAMs, and increased expression of IFN-regulated genes in DAB2 KO TAMs. Unsupervised clustering suggested thatDAB2 plays a role in macrophage differentiation, and showed the emergence of a subset among DAB2 KO TAMs with bias toward antigen presentation. Bronte and his team then validated the presence of the DAB2 protein in TAMs, but not in normal mammary tissue macrophages, and demonstrated DAB2-expressing macrophage localization along tumor borders in primary tumors as well as in metastases. Probing for the mechanism underlying higher DAB2 expression in macrophages, the group observed a decrease in DAB2 levels in cells grown in suspension, suggesting a role for mechanotransduction pathways. In silico studies showed a correlation between the YAP/TAZ pathway (involved in mechanotransduction) and DAB2 expression, and YAP/TAZ protein deletion completely abolished DAB2 expression in TAMs. In in vitro assays, DAB2-expressing TAMs promoted tumor cell invasion through haptotaxis (directional migration along a gradient of substrate-bound cues), but not chemotaxis or monocyte-associated transendothelial extravasation, and failed to do so upon DAB2 deletion. When analyzing DAB2 expression in breast cancer patients, Bronte found that the presence of high levels of DAB2+ TAMs was associated with worse prognosis. In a DAB2 KO fibrosarcoma tumor model, administration of anti-PD-1 therapy slightly reduced primary tumor growth, but significantly decreased the formation of lung metastases and led to increased NK cell activation at the metastatic site. Bronte also presented another project focusing on monocyte-induced immune dysfunction in cancer. Here, he and his team found that PD-L1+c-FLIP+ monocytes and high IL-6 serum levels correlated with poor prognosis in pancreatic ductal adenocarcinoma (PDAC) patients. c-FLIP, an anti-apoptotic protein, also demonstrated transcriptional activity upregulating several inflammatory gene signatures in monocytes, which was consistent in peripheral monocytes isolated from PDAC patients. Through further analysis of circulating CD14+ monocytes in PDAC patients, Bronte and colleagues identified CD14+ immunosuppressive monocytic myeloid-derived suppressor cells (M-MDSC) in some PDAC patients. These M-MDSCs demonstrated a distinct gene expression profile and high expression levels of STAT3 and ARG1. Bronte ended this talk by presenting his work on deciphering the state of immune dysfunction in COVID-19 patients and his contribution to improving therapy for COVID-19 patients.
Metabolic adaptation in intratumoral Tregs and its implication for cancer treatment- Ping-Chih Ho - University of Lausanne and Ludwig Institute for Cancer Research, Switzerland
Antitumor immunity depends on the infiltration and activation of tumor-specific T cells and can be attenuated by multiple tumor-microenvironmental factors, such as metabolic restrictions, hypoxia, poor T cell infiltration, co-inhibitory signals, and the presence of immunomodulatory cells such as Tregs in the tumors. While regulatory T cells suppress antitumor immunity through multiple mechanisms, including impacting T cell function and inhibiting macrophage and dendritic cell function, they also maintain immune homeostasis to prevent autoimmunity in multiple tissues. To look for ways to selectively target intratumoral Tregs, Ping-Chih Ho focused on their metabolic activity, hypothesizing that Tregs, which are present and function in many different tissues throughout the body, must be capable of adapting to available nutrients in these different tissues, including tumors. Re-analysis of published Treg RNAseq data showed upregulation of lipid metabolism-associated gene sets in intratumoral Tregs isolated from breast cancer patients compared to PBMC. Hypothesizing that enrichment of lipid metabolic pathways must be because of the increase in lipid intake, Ho and colleagues looked for the differentially expressed genes associated with lipid intake and found upregulation of CD36, a lipid translocase, as well as some lipid chaperones (FABP4 and 5) in intratumoral Tregs. Indeed, higher levels of CD36 protein were present in the tumors compared to other tissues and PBMCs of matched melanoma patients. In mice, Treg-specific CD36 deletion decreased lipid uptake by intratumoral Tregs, decreased the accumulation of intratumoral Tregs, and suppressed tumor growth in a melanoma model. Interestingly, functional suppressive activity was not impared in when CD36 was knocked out in a a colitis autoimmune mouse model. Further analysis revealed loss of mitochondrial fitness, accumulation of NADH, and upregulation of apoptosis markers in CD36 KO Tregs, making them vulnerable to tumor microenvironmental factors such as hypoxia and lactic acid accumulation. In the genetically engineered melanoma mouse model, anti-CD36 antibody treatment increased Treg apoptosis,decreased intratumoral Tregs, suppressed tumor growth, and synergized with anti-PD-1 therapy in an anti-PD-1-resistant tumor model.
Exploring the role of tertiary lymphoid structures in melanoma- Göran B. Jönsson - Lund University, Sweden
The clinical introduction of checkpoint blockades has dramatically improved outcomes for some melanoma patients. Trying to more fundamentally understand how intrinsically resistant melanomas are different from those that respond, Göran B. Jönsson focused on the role of adaptive immune cells in the melanoma microenvironment. Studying 177 melanoma immune checkpoint-naive metastases, he and this team found that 33% of the samples had CD8+ T cell infiltrates, and that 25% had CD20+ B cells present. CD20+ B cells were localized in clusters indicating the presence of lymphoid aggregates (LA) with variable degrees of “tightness”. Tumors with LAs were in all cases infiltrated by CD8+ T cells, which were mainly found outside of the LAs. Jönsson showed that LA-positive tumors had increased expression of CXCL13, CXCR5, and DC-Lamp, confirming that the LAs were tertiary lymphoid structures (TLSs) of different maturity levels. The presence of TLSs has previously been shown to be a favorable prognostic factor in many different solid tumors by boosting the antitumor immune response and, depending on the maturity level, inducing antibody-producing B cells. The survival data in this cohort of patients following ICB was stratified (best to worst outcome) based on presence of both CD20+ and CD8+ cells, CD8+ alone, or neither. Using digital spatial profiling, Jönsson and colleagues found that TLSs in the same tumor could have different maturity levels and functions, and that intratumoral B cells could be divided into two groups: Ki67low and Ki67high, whereby Ki67highPD-1highCD40highMHC-IIhigh tumor-associated B cells may operate in the germinal centers of more mature TLSs. This finding was confirmed by single cell RNAseq of two melanoma patients, confirming the presence of unswitched B cells (IgM and IgD), switched B cells (IgG), and Ki67high B cells. Tumor-associated TLSs also determined the T cell phenotype. T cells within TLSs were enriched in CD4+ T cells, while T cells from TLS-negative tumors displayed a more exhausted/dysfunctional phenotype with increased expression of immune checkpoint molecules. Looking at single cell RNAseq data, Jönsson and his team found that B cell-rich samples contained more naive/memory CD4+ and CD8+ T cells, expressing TCF7 and ILR7, compared to B cell-poor samples. The stepwise formation of TLSs in metastatic melanoma was further confirmed by Visium sequencing (used to spatially resolve the transcriptional landscape) complemented with multi-color immunofluorescence. UMAP clustering revealed ten distinct cell populations in a lymph node melanoma metastasis, including B cell and plasma cell populations. Chemokine programs that are required for TLS formation were clustered with CD20+ regions. A study by Jennifer Wargo had previously shown that memory B cells that can differentiate into plasma cells are more frequent in ICB responders, suggesting that antibody-producing B cells are crucial components of an antitumor immune response. Jönsson developed a gene signature reflecting melanomas with tumor-associated TLSs and could show that patients with high expression of this gene signature had the best survival outcome. Applying this signature to anti-CTLA-4 or anti-PD-1-treated patients predicted better survival, independent of tumor mutational burden and mutations in melanoma driver genes.
Tumor-specific T cell differentiation and dysfunction- Andrea Schietinger - Memorial Sloan Kettering Cancer Center, United States
Andrea Schietinger posited the question of what molecular mechanisms drive T cells to dysfunction over the course of tumorigenesis. Investigating this, Schietinger’s team used a genetically engineered mouse model (GEMM) of liver cancer with the SV40 large T antigen as a strong oncogenic driver, which also contains immunodominant epitopes. In this model, transferred tumor-specific T cells infiltrate into lesions, become activated, upregulate inhibitory receptors, and begin to lose function within two weeks. These effects become more pronounced over time, and while early-stage dysfunctional T cells could be reprogrammed and functionally rescued (“plastic”), late-stage dysfunctional T cells could not (“fixed”). Further, these plastic versus fixed dysfunctional states were epigenetically distinct. After identifying the transcription factor Tox as an important driver of T cell exhaustion, the team asked the question “does deletion of Tox prevent T cell exhaustion?”. When antigen-specific, Tox-deficient T cells were transferred into the same mouse model, they did not show the transcriptional or epigenetic signatures for exhaustion, but interestingly, they also did not show increased effector functions, suggesting that exhaustion programming and loss of effector functions are distinct programs that occur downstream of TCR signaling. Tox-deficient T cells also failed to persist in these models, and were enriched for activation-induced cell death (AICD) signatures. Additional data showed that chronic TCR stimulation drives NFAT to translocate to the nucleus without AP-1, driving Tox expression and ultimately leading to exhaustion programming and upregulation of inhibitory receptors, which physiologically act as a negative feedback mechanism to prevent overstimulation and AICD, and mediate T cell persistence. Finally, to further identify factors that drive phenotypic and functional heterogeneity within TILs, Schietinger’s team investigated clonal diversity by evaluating responses in T cells with TCRs that recognized the same antigen with different affinity strengths (by varying the epitope sequence recognized by a SV40 antigen-specific, high-affinity TCR) and found that T cells with high-affinity TCRs quickly produced maximum levels of IFNγ, but became dysfunctional very quickly. Meanwhile, T cells with low-affinity TCRs retained their effector functions for longer, but were unable to control tumors and so were functionally inert against cells expressing the low-affinity epitope. Interestingly, they were functional against cells expressing the high-affinity epitope, indicating they were intrinsically functional, but their response was epitope-dependent. This suggested that there may be a “goldilocks” range for optimal TCR signal strength and that fine-tuning signal strength could be used to enhance cytotoxic effects. In an effort to reduce signal strength in a T cell with a TCR signal that was too strong, the researchers modified the CD8α/β co-receptor, eliminating its ability to enhance affinity. This lowered the TCR signal strength and led to enhanced tumor control in vivo.
Remodeling the TME for tumor rejection using synthetic oncolytic nanoparticles carrying self-replicating RNA payloads- Darrell Irvine - Massachusetts Institute of Technology, United States
Inspired by how oncolytic viruses are able to initiate a self-sustaining “vaccinal cycle” (driven by immunogenic cell death [ICD] in the presence of tumor antigens), but cognizant of some of the limitations of viral-based therapy (including tissue penetration, variable cell infection, and resistance to killing), Darrell Irvine considered whether gene delivery approaches could be engineered to create a “synthetic oncolytic virus” with a similar impact. Liposomes are frequently used for nucleic acid gene delivery, and have some inherent toxicity, which is a property that could be optimized for this application. As a gene payload, Irvine chose the alphavirus self-amplifying replicon RNA, which can replicate many copies of a payload RNA within an infected cell. Three different lipids encapsulated the payload RNA efficiently, but delivery experiments into B16 melanoma cells in culture showed that the lipid known as TT3 formed nanoparticles that most effectively delivered RNA to these cells and also caused efficient immunogenic cell death (even without an RNA payload). TT3 activated TLR3 and the ISFG-3 Type I interferon signaling complex (upregulating STAT1, STAT2, and IRF9), reminiscent of innate immune activation. Initial in vivo studies following injection of TT3 particles with an mCherry-reporter gene into B16 tumors led to modest tumor control. Tumor cells were the largest transfected population and, importantly, cells began to die within 24 hours, with upregulation of calreticulin and type I interferon. Encoding the T cell-stimulating cytokine IL-12, alone or attached to the collagen-binding protein lumincan, in the replicon and intratumoral delivery led to rapid and more effective favorable TME remodeling, including infiltration of granulocytes, CD8+ T cells, and monocytes. Addition of the lumican domain was needed to prevent high levels of systemic IFNγ and associated weight loss. A single intratumoral injection of IL-12-encoding replicons were effective in the control of large, established tumors (50 mm2) in multiple models, and led to long-term survival with immunological memory. In mice bearing two tumors, untreated tumors were partially controlled in combination with anti-PD-1, and long-term survival was observed in one-third to half of the mice. Finally, to evaluate the impact of IL-12 expression on antigen-specific immune responses, cross-presenting cDC1 dendritic cells were removed from draining lymph nodes of treated mice and used to stimulate pmel T cells in vitro. IL-12-containing replicons led to the largest T cell stimulation. Tumor control effects were Batf3- and CD8+ T cell-dependent, and were reduced when components of the STING or Myd88 pathways were silenced. Altogether, these ICD-inducing, replicon-containing and IL-12 expressing nanoparticles effectively remodeled the TME leading to improved T cell immunity that synergized with anti-PD-1 therapy.
T cell therapies
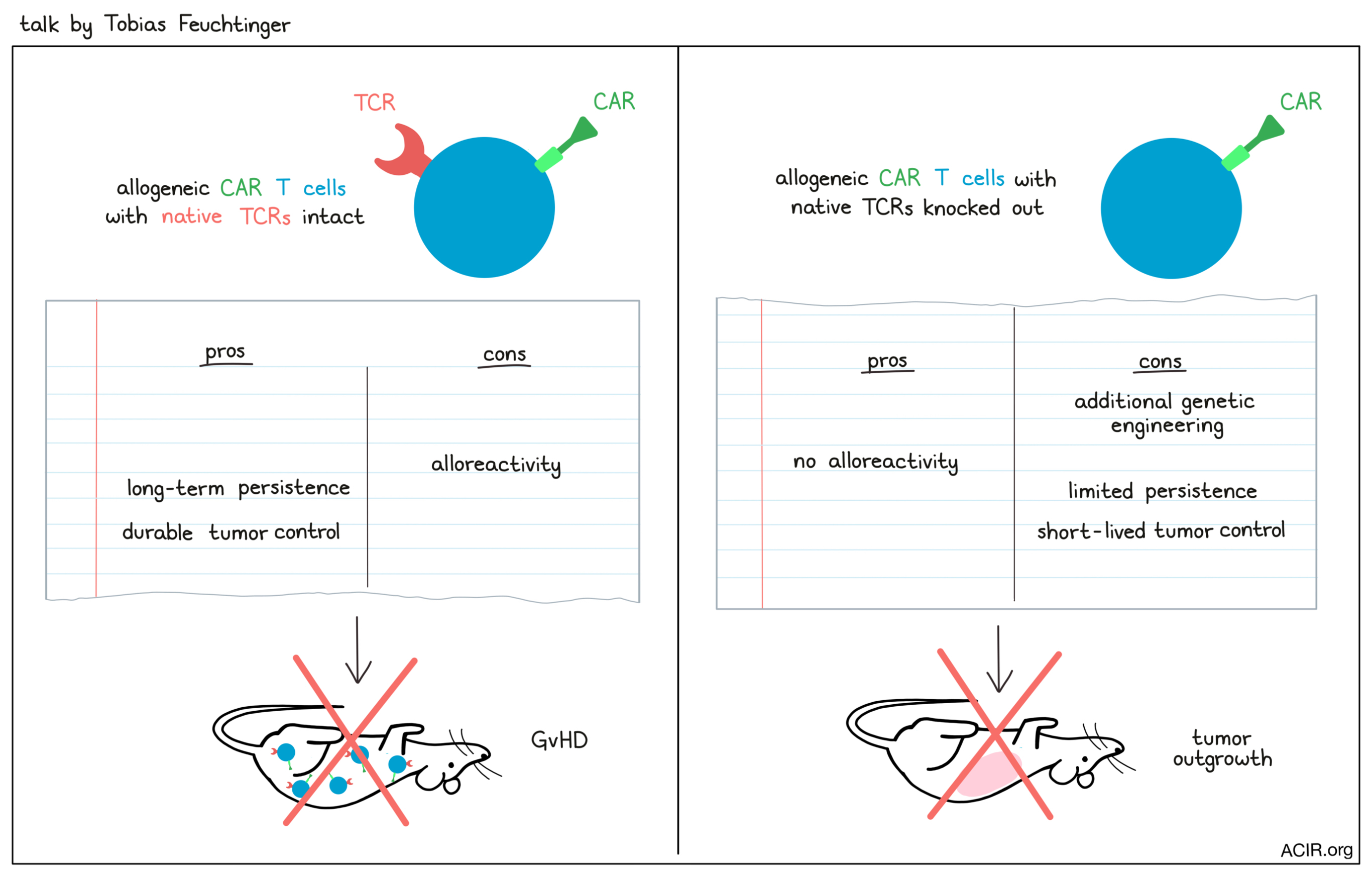
TCRs and off-the-shelf T-cell therapy?- Tobias Feuchtinger - Ludwig-Maximilians-Universität, Germany
Although anti-CD19 CAR T cells showed significant antileukemic activity in extremely ill adult and pediatric patients with B-ALL or NHL, only 50-70% of the patients who were selected for CAR T cell treatment eventually received the therapy. To overcome the challenges of time, T cell availability, and functionality in the CAR T cell manufacturing process, Tobias Feuchtinger focused on the use of allogeneic T cells. The known hurdles of such an approach are alloreactivity, immune rejection, and the challenge of engineering T cells in multiple steps under GMP conditions while maintaining functionality of the cells. To ablate alloreactivity and immune rejection of off-the-shelf CAR T cells, 3-6 engineering steps would be necessary, including depletion of TCR, MHC-I, and MHC-II expression, and, to overcome attack from NK cells, induction of Siglec ligand, HLA-E, or HLA-G expression. In a recent study, TCR knockout (KO) CAR T cells induced minimal residual disease in a patient, but a few contaminating CAR T cells still expressing a TCR expanded and caused GvHD. To prevent this from happening, Feuchtinger and colleagues developed a protocol in which they transduced peripheral T cells with a CD19 CAR, induced a CRISPR/Cas9-mediated KO of the TCRβ chain, and depleted CD3+ cells after expansion. The engineered T cells were highly functional and efficiently killed patient-derived xenograft (PDX)-grown ALL cells in vitro. However, when comparing the efficacy of TCR KO with TCR-expressing CAR T cells in NALM6 and PDX in vivo models of childhood ALL, Feutchinger and his team uncovered a dilemma. TCR KO CAR T cells initially controlled leukemia cell growth as well as TCR-expressing CAR T cells, but this control was short-lived and the leukemia rapidly returned. On the other hand, TCR-expressing CAR T cells showed superior, long-term leukemia control and persistence in vivo, but as a drawback, mediated a fatal xenoreaction (resembling alloreactivity/GvHD). Long-term disease control is a major clinical goal, and Feutchinger cautioned that for allogeneic donor-derived CAR T cells therapies, the long-term relevance of the endogenous TCR and the consequences of the multiple engineering steps needed to generate such cells are still notable issues.
Targeting leukemia with T cell receptors from healthy donors- Johanna Olweus - Institute for Cancer Research, Oslo University Hospital Radiumhospitalet and the University of Oslo, Norway
CAR T cell therapy targeting self antigens with cell type-restricted expression can cure patients with B cell malignancies, but translation of this success to other tumor types has been limited by availability of targets. Johanna Olweus hypothesized that using TCRs instead of CARs would dramatically increase the number of candidate antigens by including intracellular targets and would enable this translation. High-affinity TCRs that recognize self antigens are depleted to establish immunological self-tolerance, but in the setting of a tissue transplant, once immune suppression is removed, allogeneic T cells positively selected based on different HLAs can recognize normal cells from another individual expressing foreign HLA and reject the tissue. Olweus and her team found that, in contrast to the dogma that transferred T cells mainly recognize the foreign HLA molecule itself, T cells can reject a single cell type by recognition of tissue-specific peptides presented on foreign HLA. They identified two donor-derived TCRs that recognize epitopes derived from the terminal deoxynucleotidyl transferase (TdT) in the context of foreign HLA-A02:01. TdT is a critical enzyme in TCR and BCR rearrangement, and is highly specific for differentiating T and B cells, but is not expressed in hematopoietic stem cells, naive or mature T or B cells, or plasma cells. TdT is also homogeneously overexpressed in 80-90% of B-ALL and T-ALL. T cells engineered to express the identified TCRs showed high peptide specificity and were dependent on HLA-A2. TdT TCR T cells did not respond to a panel of HLA-A2+ cell lines that expressed a broad repertoire of additional HLA-alleles, unless these cells were loaded with TdT peptides or expressed TdT. Olweus and colleagues then used bioinformatic and cellular approaches to study the fine specificity of the candidate therapeutic TCR and identify potential cross-reactivity to normal peptides in the normal human proteome by interrogating all possible single amino acid changes in the target peptides. They found that amino acid-substituted hit sequences did not match any natural peptide in the human proteome. In vivo, TdT TCR T cells mediated rejection of human leukemia cell lines (BV173 or NALM-6) and primary B-ALL to negative minimal residual disease levels. Coculture of TdT TCR T cells with primary patient-derived B-ALL or T-ALL cells showed killing of leukemic cells, but sparing of normal B cells, T cells, and CD34+ cells. Using colony-forming assays and a humanized mouse model, Olweus and her team could not detect TdT TCR T cell toxicity on normal human hematopoiesis or human thymocytes, which, in general, do not co-express HLA-A2 and TdT at high levels. In conclusion, Olweus and colleagues have developed a novel adoptive T cell therapy approach that could be used to treat patients with relapsed CD19- B-ALL (after CD19 CAR T cell therapy) or relapsed T-ALL expressing high levels of TdT.
Engineering next-generation T cells for cancer immunotherapy- Yvonne Chen - University of California Los Angeles, United States
Antigen escape significantly limits durability of response to CAR T cell therapy, so Yvonne Chen tried to overcome this problem by varying the CAR design. She and her team have recently developed bispecific ‘OR-gate’ CD19-OR-CD20 CAR T cells. In an ongoing phase 1 dose-escalation clinical trial initiated in October 2019 in CLL and NHL patients, 4 out of 5 evaluable patients (80%) have achieved a complete response to treatment with CD19-OR-CD20 CAR T cells and have remained in complete remission since then (4–15+ months). No neurotoxicity and no cytokine release syndrome above grade 1 have been observed. One patient with 4 prior lines of treatment, including rituximab and two other immunotherapy clinical trials by VelosBio (targeting ROR1) and Amgen (anti-CD19 BITE), showed at the initial screening (in August 2020) the presence of CD19dim or CD19- tumor cells that continued to express CD20. CD19-OR-CD20 CAR T cells retain full efficacy even when only one antigen is present, and this patient, despite near antigen loss of CD19, achieved complete remission that is now ongoing for more than 6 months. Chen and her colleagues applied their expertise to create another true OR-gate CAR that targets CS1 and BCMA for the treatment of multiple myeloma. For the in vivo validation of this CAR, mice were engrafted with a heterogenous population of BCMA+/CS1+, BCMA-/CS1+, and BCMA+/CS1- multiple myeloma cells. The OR-gate CAR T cells outperformed single-input CAR T cells and persisted in the mice, but were not able to control tumor growth long term. Persistent CAR T cells expressed high levels of PD-1, and combination therapy with anti-PD-1 improved the initial antitumor efficacy, but by day 130, there was no difference in overall survival between groups. Surprisingly, animals treated with the combination actually did substantially worse upon tumor rechallenge. BCMA-OR-CS1 CAR T cells recovered from animals treated with anti-PD-1 expressed high levels of CD38, indicating that CAR T cells that are exposed to anti-PD-1 during a period of low antigen stimulation following tumor clearance may become dysfunctional. In an ongoing study that is part of early clinical development, Chen and her team are stopping anti-PD-1 treatment shortly after clearance of the initial tumor burden to prevent CAR T cell dysfunction. These researchers have also developed CAR T cells that can not only resist immunosuppressive TGFβ, but can also convert high TGFβ (present in the tumor microenvironment of solid tumors) to an activating signal via an intracellular EGFR signaling domain. The proliferative activity of the TGFβ CAR T cells is dose-dependent and ensures stimulation of the CAR T cells only in the tumor microenvironment, and only by the active, but not the latent form of soluble TGFβ. The TGFβ CAR itself does not confer cytotoxic activity, but was incorporated into a bispecific IL-13Rα2/TGFβ CAR. In a solid, orthotopic tumor model with engrafted patient-derived GBM neurospheres, IL-13Rɑ2/TGFβ CAR T cells exhibited superior control.
Neoantigens and cancer vaccines
Capturing heterogeneity and the HLA-presented landscape in melanoma- Yardena Samuels - Weizmann Institute of Science, Israel
Tumor-infiltrating T cells secrete IFNγ, which induces indoleamine 2,3-dioxygenase 1 (IDO1) to catalyze the conversion of tryptophan into kynurenine, a known suppressor of T cell function. Knowledge of this pathway led to the hypothesis that inhibition of IDO1 would rescue kynurenine-mediated T cell suppression. However, several clinical trials reported lack of efficacy of IDO1 inhibitors in combination with PD-1 pathway blockade. Running a lab focused on understanding the HLA:peptide::TCR interface, Yardena Samuels turned to the impact of uncharged (tryptophan) tRNAs on peptide translation within the cancer cell to find clues to explain this failure. Samuels began by analyzing Ribosome Protected Fragments (RFPs) and transcriptome data in 3 melanoma cell lines following prolonged exposure to IFNγ (which stabilized IDO, decreased tryptophan, and increased kynurenine as expected). Samuels and her team noticed that in IFNγ-treated cells, in addition to the expected accumulation of RFPs due to ribosome pausing at translation start sites, there was also ribosome pausing on Tryptophan (W) codons (W-bumps), and then continuation of translation with additional pausing and accumulation of RFPs (W-bumps) 15 amino acids after the Trp codon, which was IDO1-dependent. Drawing inspiration from ribosomal frameshifting in bacteria cultured under amino acid-starved conditions, Samuels and her team used gene constructs to demonstrate IFNγ-induced ribosomal frameshifting events in cancer cells, as well as the specificity for only IFNγ-sensitive, but not IFNγ-resistant cancer cells, in antigen-specific CD8+ T cell cocultures. IFNγ-induced ribosomal frameshifting was IDO1-dependent and led to the production of over 120 different frameshifted peptides, detected by deep protein mass spectrometry. To assess if these aberrant peptides are presented on the cell surface, HLA peptidomics was performed on IFNγ-induced and Trp-depleted cells, and multiple frameshifted peptides presented on HLA class I were identified on both cancer cell lines from a patient, as well as on metastases derived from same patients. The immunogenicity of these HLA-presented aberrant peptides were then tested for their ability to prime naive CD8+ T cells from healthy donors. Out of 180 tetramers-sorted T cells, 13 KCNK6 peptide-reactive T cell clones were grown, which were reactive to KCNK6 peptide-loaded K562-B07:02 cells. To summarize, infiltrating T cells produce IFNγ, which activates IDO1 in the cancer cells, leading to conversion of tryptophan into kynurenine and suppression of T cell function. Tryptophan depletion, at the same time, causes ribosomal pausing at tryptophan residues, and formation of W bumps, producing frameshifted peptides, which can be immunogenic. Treatment with IDO inhibitors might then reduce the utility of any such frame-shift-specific T cells that might be present. Samuels also introduced another project in which her group, for the first time, showed microbial-derived HLA-bound peptides in melanoma. Samuels and colleagues identified bacterial populations in metastatic tumors using 16S ribosomal DNA sequencing, and then performed HLA peptidomics to identify bacteria-derived peptides using the proteomes of the identified bacterial population. Bacteria-derived peptides showed higher hydrophobicity and were capable of inducing immunoreactivity.
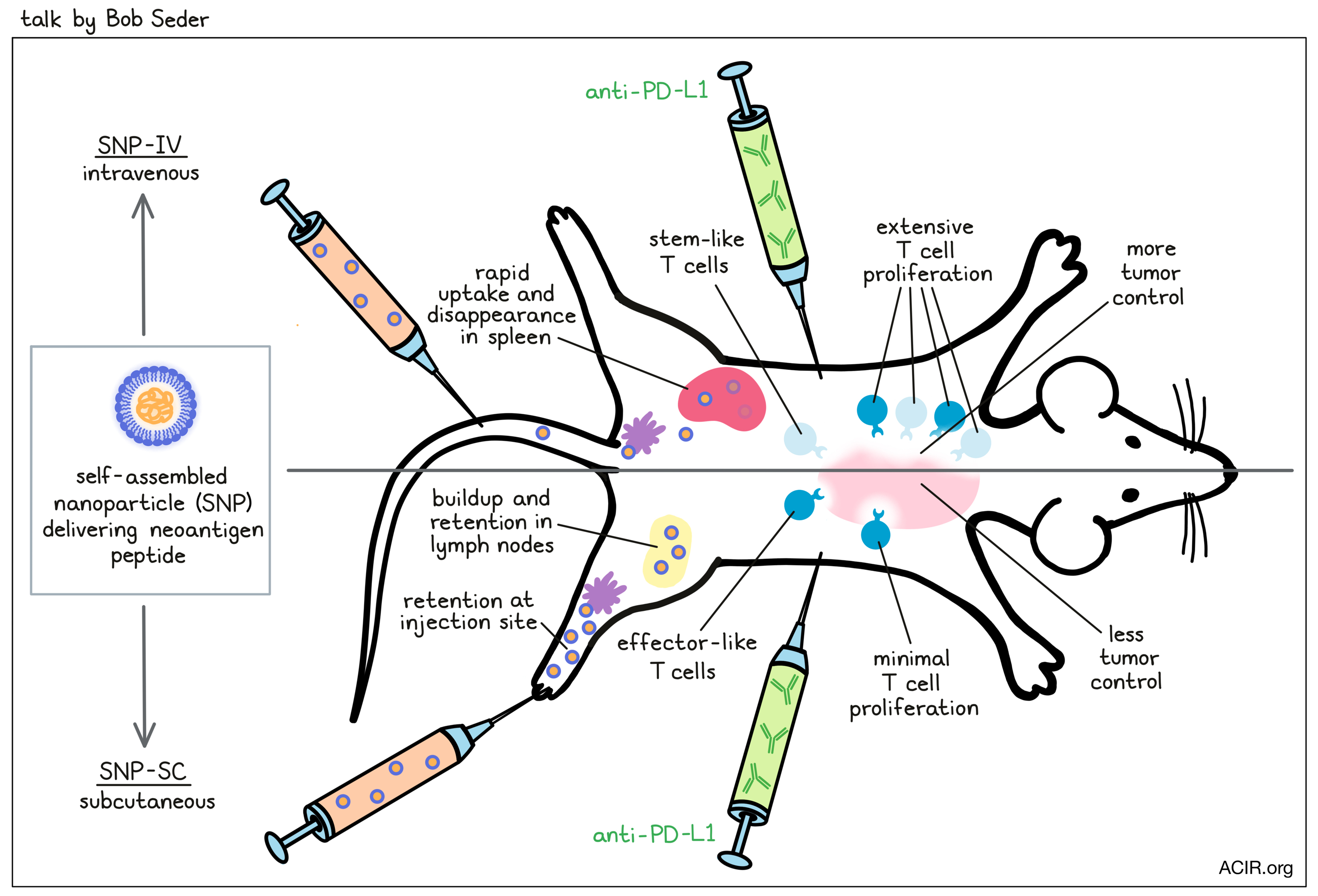
Systemic delivery of a nanoparticle cancer vaccine generates effective antitumor immunity- Robert Seder - NIH-NIAID, United States
Based on the accumulated data over the past years that checkpoint responsiveness is linked to tumor mutation burden, and hence neoantigen load, and that adoptive TIL therapy also has a neoantigen focus, Bob Seder described their efforts to improve peptide vaccine delivery to optimize quantity and quality of neoantigen-specific T cells. Based on their initial observation that for previously evaluated MC38 long peptide antigens, even for epitopes detected by mass-spectrometry, the immunogenicity of multiple peptides positively correlated with insolubility, Seder and colleagues proactively incorporated peptides into a viral-mimicking, “insoluble” particulate vaccine complete with co-delivered adjuvant. Peptides were modified during synthesis to be flanked by cathepsin cleavage sites and a solubilizing sequence, and were formulated with a TLR-7/8a-containing polymer backbone to produce micellar-like, 50–100nm self-assembling nanoparticles (SNP-7/8a). In mice, the nanoparticle vaccine delivered subcutaneously (SNP-SC), increased both the breadth of epitopes that generated a response and the magnitude of response. Interestingly, compared to delivering the particle intravenously (SNP-IV), SNP-SC generated stronger responses, even at lower doses. However, a 10x lower magnitude of SNP-IV-induced T cells together with anti-PD-L1 prevented tumor growth to an extent that was comparable to SNP-SC-induced T cells with or without anti-PD-L1, suggesting that the T cell quality was different. Moreover, vaccination in a therapeutic setting, in which doses were adjusted so each delivery route resulted in ~10% antigen-specific T cells, demonstrated improved control in the SNP-IV-vaccinated mice, which was again dependent on boosting and anti-PD-L1. Considering that this may be due to a more stem-like phenotype of SNP-IV-induced T cells, the team conducted single-cell RNAseq after prime/boost immunization of tumor-naive animals and observed expression of markers consistent with TSCM (TCF1, slamf6, Xcl1, Tox, CTLA-4), while SNP-SC-induced T cells were more effector-like (GZMB, Klrg1, CX3CR1). Kinetically, after priming of tumor-bearing animals, the frequency of TCF1+ cells was ~2x higher in SNP-IV-vaccinated mice, and after boosting and administration of anti-PD-L1, the number of stem-like cells dropped less and the number of effector cells increased more compared to mice that either received the boost without anti-PD-L1 or just anti-PD-L1. Mechanistically, the SNP-SC vaccine first concentrated in the subcapsular region of the draining lymph node and persisted for up to 14 days, slowly moving into dendritic cell zones, while the SNP-IV vaccine quickly spread throughout the body, was eliminated quickly (within a day), and appeared in monocyte-derived dendritic cells in the spleen for only about 1 day, suggesting that the transient burst of vaccine exposure might be relevant to the induced T cell phenotype. Both Batf3-dependent cDC1s and monocyte-derived DCs appear to be critical antigen-presenting cells involved in modulating the T cell phenotype for this nanoparticle vaccine.
Neoantigens beyond SNVs: embracing the full spectrum of antigenic dark matter in tumors- Alex Rubinsteyn - University of North Carolina, Chapel Hill, United States
Although many investigators are aiming to improve personal cancer vaccines with various approaches, Alex Rubinsteyn posited that a fundamental missed opportunity to date is broadening the types of neoantigens that can be evaluated due to the limitations of the current whole-exome sequencing (WES) and RNA sequencing methods using the Illumina short-read sequencing technology that is the mainstay of most pipelines. WES leads to a preponderance of single-nucleotide variants (SNVs) and very small indel mutations, missing larger indels, MSI frameshifts, gene fusions, inversions, etc., and Rubinsteyn showed data that even for SNVs, low-coverage whole-genome sequencing was more sensitive than higher-coverage WES. Similarly, current short-read RNA sequencing approaches miss large transcriptional events, such as alternative splicing, and detection of fusions can be insensitive. In particular, the ability to capture more frameshifting events can lead to longer, potentially more impactful neoepitope regions. A solution is application of longer-read sequencing based on other sequencing platforms, such as Oxford Nanopore or PacBio, both of which have been shown to significantly expand the number of detected and actionable mutations, including many types of events that are totally missed with current short-read technology. Rubinsteyn’s lab is currently running a “bake-off”, comparing different sequencing technologies on a range of disease samples to fully characterize the potential of mutations beyond SNVs, with the near-term target of conducting a clinical trial based on epitopes predicted from the ‘large’ mutations that can be uniquely detected with this newly developed neoantigen identification pipeline.
Personal neoantigen vaccines in patients with melanoma: long-term responses and combination with PD-1 inhibitor- Patrick Ott - Dana-Farber Cancer Institute and Harvard Medical School, United States
In 2017, Patrick Ott et al. and Ugur Sahin et al. published back-to-back papers describing the results of their personal neoantigen vaccine studies. Ott began his presentation by outlining both the strong rationale and excitement for targeting personal neoantigens, and the early results from the 6 patients in their study. After a median follow-up of 2 years, the vaccine had been shown to induce strong CD4+ and detectable, but weaker CD8+ T cell responses to over 60% of the immunizing peptides, some of which showed reactivity to autologous tumors. The vaccines also showed hints of clinical utility in two patients who relapsed shortly after vaccination, but demonstrated complete responses after a short course of anti-PD-1 treatment. Similar results were observed by Sahin et al. using an mRNA-based vaccine. Ott and his team followed their initial patients for a median of 4.5 years after vaccination, and added two more patients. All 8 patients are still alive, and 6 have no evidence of disease. Analysis of T cells in these patients 2 to 4.5 years after vaccination demonstrated that the majority of initial responses were durable. Single-cell RNA sequencing of neoantigen-specific, tetramer-sorted CD4+ T cells ex vivo from 4 patients over the course of vaccination demonstrated a phenotypic shift from cytotoxic effector and antigen-induced cell death signatures during priming, to a memory phenotype four weeks after the last boost. Single-cell TCR sequencing of tetramer-sorted CD4+ T cells, and bulk RNA sequencing of tumor and PBMC samples revealed a dynamic change in clonotypes during vaccination, long-term persistence of multiple clonotypes, and infiltration of multiple vaccine-induced clonotypes into a recurrent tumor that appeared shortly after vaccination (a tumor that disappeared following anti-PD-1 therapy). Finally, Ott presented evidence for on-target cytotoxic activity of the vaccine therapy based on detection of epitope spreading to non-target neoantigens and tumor-associated antigens, apparent as early as 16 weeks after initiation of vaccination. Ott then also described results from a larger, single-arm, industry-sponsored Phase Ib trial in patients with metastatic melanoma, bladder cancer, or NSCLC, in which the peptide vaccine was given 12 weeks after initiation of an up-to-two-year course of anti-PD-1 therapy. This trial featured an extensive collection of tumor and peripheral blood biopsy samples that allowed extensive immune monitoring. Analysis of T cell responses demonstrated results comparable to the earlier trial, including durability of many responses (measured at 7 months after the last boost), trafficking of a vaccine-induced CD8+ T cell to the tumor, and epitope spreading. Clinically, the reduction in tumor burden was in line with or slightly better than historical controls. Histopathology of tumor biopsies before, during, and after vaccination showed major pathological responses in some patients, occurring following initiation of anti-PD-1 therapy, but before vaccination, and responses only after vaccination in most patients, with evidence for improved progression-free survival in those patients with a pathological response.
Microbiome
Microbiota-centered interventions in cancer- Laurence Zitvogel - Gustave Roussy, INSERM, University Paris Saclay, France
Laurence Zitvogel focused her talk on the impact of the gut microbiome on cancer immunotherapy. She highlighted the meta-analysis of over 11,000 patients in 38 studies who showed poor prognosis after having received antibiotics two months prior to or during initiation of immune checkpoint inhibitor (ICI) therapy. Other similar studies further showed that in addition to antibiotics, steroids and proton pump inhibitors also dampened the effects of ICI, while chemotherapies did not. Additionally, the microbiome richness played a positive role, while some particular species played a negative role in the prediction of the therapeutic success. Zitvogel emphasized the results of two clinical trials of fecal microbiota transplantation (FMT). In both studies, FMT from patients with complete responses to immunotherapy were transplanted to immunotherapy-refractory patients and led to objective responses in a subset of patients, with colonization of fecal microbiota and favorable changes in immune cell infiltrates. Gut microbiomes impacted immunotherapy through multiple mechanisms, including tumor-intrinsic effects such as chemotaxis or antigenicity, as well as host-intrinsic effects such as epithelial barrier integrity and TME reprogramming. At the molecular mechanism level, Zitvogel further highlighted the work of Yardena Samuels that identified microbial-derived HLA-bound peptides (see above), and recent work from her own lab showing that Enterococcus phage harbors antigenic MHC class I-restricted peptides that share similarities with tumor antigens; the T cell response directed against this phage can eliminate cancer. At the bacterial species level, analysis of fecal samples from 100 patients with kidney and lung carcinoma had previously shown that the fecal composition at the start of the ICI therapy could influence the therapy response, and that the prevalence of Akkermansia was higher in responders. In a prospective validation cohort of 466 NSCLC patients receiving anti-PD-1 therapy, Akkermansia muciniphila (Akk) was prevalent in patients achieving a complete or partial response after anti-PD-1 therapy, and was associated with improved overall survival. However, Akk+ stools also showed increased microbial diversity. Interestingly, in a closer look at a cohort of 368 patients with no, low, or high levels of Akk, patients with low levels of Akk had the best prognosis, while negative and high levels of Akk were associated with poor OS. In addition, Akk levels dictated the taxonomic composition of the patients, with Akklow patients enriched for microbiota associated with better prognosis, such as Lachnospiraceae and Ruminococcace, and favorable tumor microenvironments with higher levels of CD3, INFγ, CXCL10, and ZBP1. Akk levels also predicted responses to PD-1 blockade better than PD-L1 levels or antibiotic treatment. Transfer of Akkneg, Akklow, or Akkhigh human feces from NSCLC patients into tumor-bearing avatar mice showed that Akklow fecal transfer mediated the most tumor control with anti-PD-1 therapy. The exogenous administration of a lyophilized version of Akk only benefited the mice with Akkneg fecal composition, suggesting that Akkneg patients might benefit the most from lyophilized Akk before immunotherapy.
Manipulating the gut microbiome to improve current immunotherapies of cancer- Hassane M. Zarour - UPMC Hillman Cancer Center, United States
Following Laurence Zitvogel’s talk, Hassane Zarour further emphasized the multiple reports of the gut microbiome composition in modulating anti-PD-1 therapy effectiveness, but also highlighted discrepancies and lack of commonality among studies. In their own study of 63 patients with melanoma who were treated with anti-PD-1 therapy, baseline fecal microbiome composition was the best predictor of progression-free survival at 9-10 months, with several previously identified beneficial and detrimental taxa associated with response to anti-PD-1 therapy outcome. To understand the reasons behind the lack of commonality in identified microbiota strains in different studies, Zarour and colleagues performed a meta-analysis comparing their data with those of previously published studies using a single analysis pipeline. They found geographical locations of the studies, tumor types, and sequencing technologies used in the studies affected the microbiota identification. Further analysis of 16S datasets from the human microbiome project identified 27 clusters of commensals, and their distribution varied based on geographical locations, suggesting that the variations observed in different studies may be due to the geographical differences. Mapping the published data from melanoma patients based on responders and non-responders, Zarour and colleagues identified 4 superclusters of microbiota: Beneficial 1 and 2, and Detrimental 1 and 2. These clusters varied in microbiota composition, but were consistent with other studies. Further mapping the superclusters based on locations showed an abundance of beneficial supercluster 1 in Pittsburgh and Chicago, and an increased amount of beneficial supercluster 2 in New York and Houston. Arguing that enough information was not yet available to pick particular species for microbiome modulation, Zarour then introduced the results of phase 1 clinical trial of fecal microbiota transplant (FMT) from patients who responded to anti-PD-1 immunotherapy into patients who were refractory to anti-PD-1. FMT-treated patients were continued on anti-PD-1 therapy. Out of 15 refractory patients, 1 achieved CR, two achieved PR, and 3 had stable disease, with responders showing a more consistent shift toward the donor composition. FMT also altered the specific fecal microbiota composition, with increases in Actinobacteria and Firmicutes and decreases in Bacteroides and Proteobacteria in responders. FMT increased intratumoral and systemic immunity in the responders, with increases in antibodies targeting donor microbial species, activation of CD8+ T cells and MAIT cells, and decreases in Tregs. Increases in a myeloid cell gene signature associated with melanoma progression were observed in non-responders. Responders showed distinct circulating cytokine and chemokine signatures and metabolomic signatures. Transkingdom network analysis further suggested that microbiome compositions mediated immunological, cytokine/chemokine, and metabolomic changes in responders.
By Ute Burkhardt, Ed Fritsch, Lauren Hitchings, and Shishir Pant



