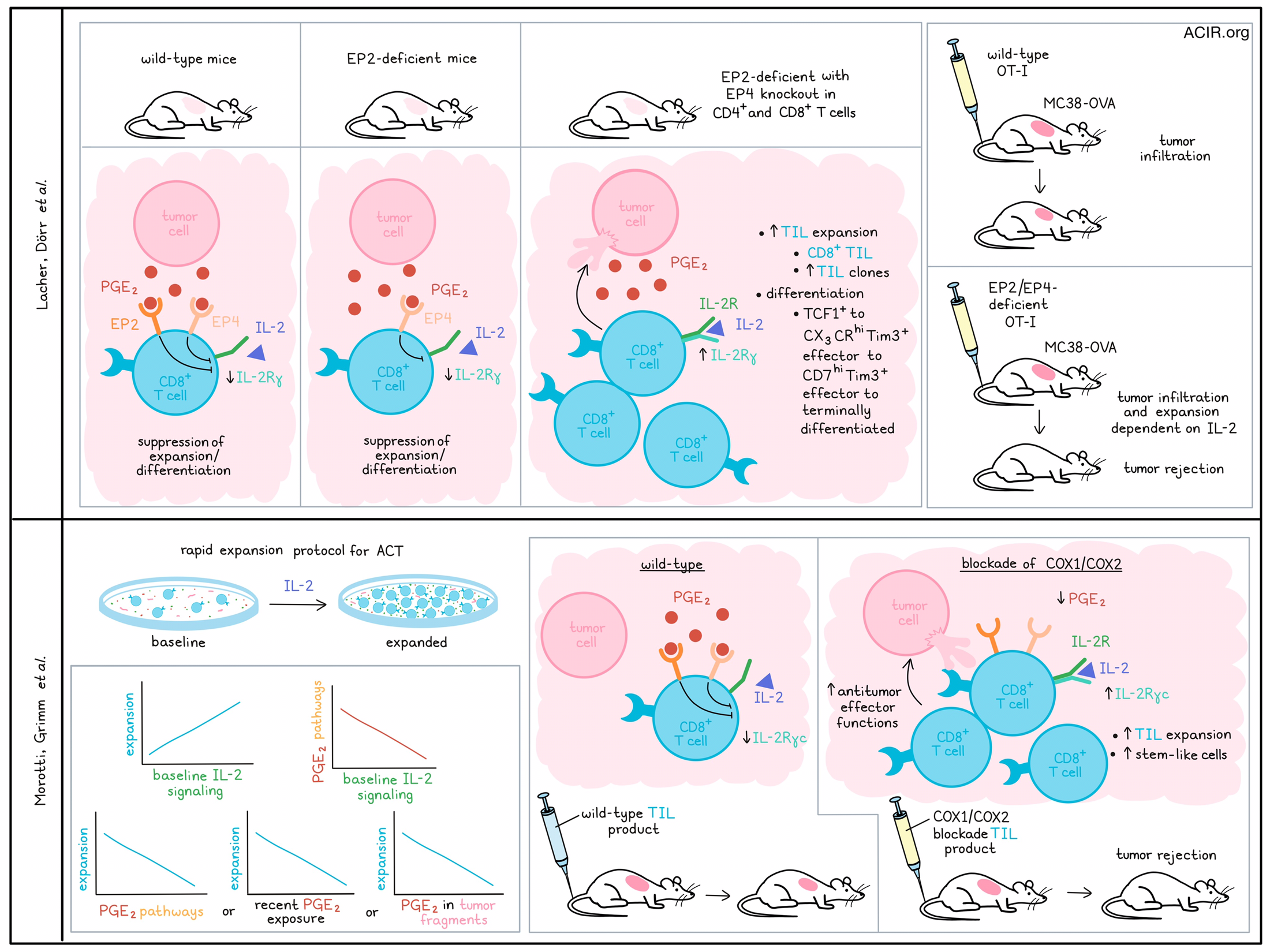
The bioactive lipid prostaglandin E2 (PGE2) is often detected in the tumor microenvironment (TME), and has been shown to play a role in immune escape and cancer progression. Two recent studies published in Nature assessed the effects of PGE2 on tumor-infiltrating lymphocytes (TIL). Lacher, Dörr, et al. investigated its impact on TCF1+CD8+ T cells in mice, while Morotti, Grimm, et al. assessed how PGE2 affects ex vivo TIL expansion for adoptive cell transfer (ACT) in humans.
PGE2 signaling is mediated by four receptors expressed on immune cells, of which two, EP2 and EP4 (encoded by Ptger2 and Ptger4 in mice), are known to suppress immune function. Therefore, Lacher, Dörr, et al. created a mouse model (Cd4crePtger2-/-Ptger4fl/fl) in which mice were germline EP2-deficient, and EP4 was deleted in CD4+ and CD8+ T cells; control mice were either WT mice or lacking only in EP2. The Cd4crePtger2-/-Ptger4fl/fl mice rejected PGE2-producing BRAFV600E melanoma tumors, while tumors grew in WT mice and in mice lacking only EP2. The tumor rejection in the Cd4crePtger2-/-Ptger4fl/fl mice was associated with an increase in CD8+ but not CD4+ TIL, suggesting the differences in rejection were due to CD8+ T cell-related effects.
scRNAseq and scTCRseq on CD8+ TIL from BRAFV600E tumors revealed 8 TIL clusters, which all expressed Pdcd1 (PD-1), Cd44, and Tox. Clusters 1 and 2 highly expressed stem-like TCF1+ T cell markers (Tcf7, Slamf6, and Il7r). Cluster 1 TCF1+ TILs expressed Sell (CD62L), Ccr7, and Bach2, while cluster 2 TCF1+ TILs expressed effector function and migration markers. The other clusters lacked Tcf7, and expressed genes associated with T cell differentiation and effector function, including Havcr2 (Tim3) and Cxcr6, suggestive of early and/or terminally differentiated TIL phenotypes. Unsupervised slingshot analysis showed a developmental trajectory from TCF1+ cells that progressed to CX3CR1hiTim3+ effector cells, then into CD7hiTim3+ effector cells, after which cells progressed to terminally differentiated subsets. In Cd4crePtger2-/-Ptger4fl/fl mice, there were highly expanded TIL clones, higher proportions of clones shared between TCF1+ cells and Tim3+CXCR6+ effector cells, and a shift towards early effector and terminally differentiated TIL populations. Mice only lacking EP2 signaling had no expansion of any differentiating effector TIL populations. The data suggested that PGE2 prevented differentiation and expansion of effector T cells that arise from TCF1+ TILs, and EP2/EP4 deficiency reversed this blockade.
Treatment with FTY720, which prevents newly-primed T cells from moving out of the lymph node, beginning on day 6 did not impact the intratumoral formation and expansion of Tim3+ TILs. However, these effects were blocked when FTY720 was used from day 1, suggesting that TCF1+ TIL locally generated effector responses when PGE2 signaling was blocked.
EP2/EP4 deficiency in TCF1+ TIL increased the expression of transcription factors related to effector differentiation, stimulatory cytokine signaling, and survival. This included increases in transcription factor activity related to IL-2 signaling, which led the researchers to hypothesize that PGE2 might impact the effects IL-2 has on TILs. In vitro experiments with TCF1+ TILs sorted from PGE2-deficient tumors showed that their expansion and differentiation into effector cells in response to IL-2 and CD3/CD28 stimulation was limited by PGE2. Further assessing these effects in antigen-experienced, repeatedly-activated (RA) TCF1+CD8+ T cells generated in vitro with RNAseq showed PGE2 treatment induced transcriptional changes, with increases in EP2/EP4-mediated cAMP signaling and T cell quiescence. Cells also downregulated T cell differentiation-associated mTORC1 and the IL-2 signaling pathway. Further, IL-2 stimulation of these PGE2 cells did not induce STAT5 phosphorylation and the cells had reduced expression of the IL-2 receptor γ-chain, indicating a dominant inhibitory effect of PGE2.
Morotti, Grimm, et al. were also interested in factors affecting the T cell response to IL-2 and investigated how the TME affects the response to IL-2 in human TILs during the rapid expansion protocol for ACT. scRNAseq and scTCRseq analyses of baseline and expanded TILs from patients with melanoma allowed for assessing the baseline TIL state and the subsequent response to IL-2 rapid expansion. There was an association between the baseline IL-2 signaling gene signature and TIL expansion. Assessing TME factors that may impact IL-2 responses in the in vitro cultures, the researchers found an inverse correlation between IL-2 signaling and PGE2-linked pathways.
To determine which TIL clonotypes could sense PGE2 in tumors, a gene signature for recent PGE2 exposure in CD8+ effector T cells was developed using an in vitro culture system with RA T cells. This signature was detected in tumor-reactive CD8+ TILs in baseline tumors, and its expression correlated with poor ex vivo expansion in response to IL-2, in particular in those CD8+ TIL that had a precursor-exhausted, terminally exhausted, or effector memory-like phenotype. Additionally, high levels of PGE2 in tumor fragments in TIL cultures correlated negatively with in vitro TIL expansion. EP2/EP4 blockade with small molecule antagonists reversed the inhibitory effect of PGE2 on TIL expansion.
Assessing how PGE2 negatively impacts the action of IL-2 on TIL, the researchers, similar to Lacher, Dörr, et al., found reduced surface expression of IL-2Rγc on TIL in response to PGE2, which was reversed by EP2/EP4 blockade. Additionally, exposure to PGE2 reduced the surface colocalization of IL-2Rβ and IL-2Rγc in TILs. Stimulation of PGE2-exposed RA T cells with exogenous IL-2 did not induce downstream signaling of IL-2Rγc (AKT, mTOR, S6), and this was also true when an IL-2 variant (IL-2v) that selectively binds to IL-2Rγc was used.
Bulk RNAseq of unstimulated and RA CD8+ T cells after PGE2 exposure revealed an increase in gene expression related to protein kinase A signaling, cAMP-dependent signaling, and metabolic processes. In RA CD8+ T cells, PGE2 inhibited several IL-2-mediated pathways associated with T cell proliferation and regulation of metabolism, leukocyte proliferation, and the mTOR pathway. Also, PGE2 induced transcriptional changes in RA CD8+ T cells related to mitochondrial and lipid metabolism, as it upregulated genes associated with dysfunctional exhaustion, cell cycle arrest, mTOR inhibition, lipid metabolism, and ferroptosis. There was also increased oxidative stress, mitochondrial dysfunction, and a lack of nucleotide synthesis.
Morotti, Grimm, et al. then assessed whether PGE2 blockade during TIL expansion protocols could restore the TIL response to IL-2. Blocking EP2/EP4 receptors in TIL increased the number of expanded TILs. Similarly, blocking COX1 and COX2 reduced PGE2 in the culture medium, increased TIL expansion, and increased IL-2Rγc expression on TIL. Further, it increased the TILs’ mitochondrial fitness and the expression of TCF7 and MYB, which are related to stem-like T cell longevity. Overall, COX inhibition resulted in more proliferation-competent precursor-like TILs in the culture, as there was an increase in stem-like CD39-TCF1+ cells and a decrease in TOX+TCF1- cells. Further, these effects seemed preferentially induced in tumor-reactive TILs, as they upregulated CD137 and IFNγ/TNF production in autologous in vitro tumor coculture assays.
To test whether this translated into better tumor control by the TIL product, TILs were transferred into NSG mice together with patient-derived tumor cells. Control cells could not control tumor growth, but the COX inhibitor-treated TILs induced tumor rejection. In NSG mice with patient-derived xenograft melanomas, these TIL were more abundant intratumorally and had higher CD103 and lower PD-1 expression.
Finally, Lacher, Dorr, et al. also tested the effects of blocking PGE2-EP2/EP4 signaling on tumor growth. WT or EP2/EP4-deficient OT-I T cells were transferred into mice challenged with MC38-OVA tumors. Both WT and EP2/EP4-deficient OT-I cells induced similar levels of TCF-1+ cells, indicating that lymphoid tissue priming was not impacted, but WT T cells halted expansion after tumor infiltration, while the EP2/EP4-deficient OT-I T cells persistently expanded in the TME. These effects were inhibited when IL-2R signaling was blocked, suggesting IL-2R signaling drove the expansion and differentiation of antigen-specific CD8+ TILs when PGE2-EP2/EP4 signaling was absent. When transferred into WT mice, these EP2/EP4-deficient OT-I cells induced complete rejection of MC38-OVA tumors.
Together, these studies show that tumoral PGE2 negatively impacted TIL expansion and effector differentiation. Blockade of the PGE2-PE2/PE4 pathway in the tumor may help induce better T cell antitumor responses. Given its effects on IL-2 response by the TIL, PGE2 might be an important therapeutic target for both IL-2-directed therapies (such as IL-2v) and TIL manufacturing.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Sebastian Lacher answered our questions.

What was the most surprising finding of this study for you?
One of the most astonishing findings we made was how PGE2 affected CD8+ T cells in tumors. Contrary to our expectations, PGE2 did not seem to induce a dysfunctional gene expression program or promote T cell exhaustion, nor did it show a significant impact on the ability of cytotoxic CD8+ T cells to execute effector functions. Instead, PGE2 mediated its deleterious effect by throttling T cell expansion and effector differentiation from tumor-infiltrating stem-like CD8+ T cells – a mechanism that seems to be rather unique compared to other previously known inhibitory mechanisms limiting T cell immunity in cancer. A second astonishing finding was how effective tumor-infiltrating CD8+ T cells could mount anti-cancer responses and eliminate tumors when the T cells lacked the PGE2 receptors EP2 and EP4.
What is the outlook?
We hope that our findings may help to improve immunotherapies that are based on targeting T cells or using patient-derived T cells. A companion paper by Morotti & Grimm et al. (2024) already provides evidence that this is indeed the case by showing that PGE2 undermines the activity of human TILs and their use for adoptive T cell therapy (ACT). One of our key findings was that PGE2 impaired IL-2 responsiveness in TILs; therefore, it will be intriguing to see whether blocking the PGE2-EP2/EP4 axis can promote better responses to current immunotherapies using IL-2. Moreover, it will be exciting to see whether blocking PGE2 signaling will play a substantial role in therapies such as chimeric antigen receptor (CAR) T cell or antibody–drug conjugate therapy.
What was the coolest thing you’ve learned (about) recently outside of work?
During my vacation in Panama, my American friend and I went on a snorkeling trip to see the stunning marine life. While exploring Punta Laurel, I swam to a recommended spot with my underwater camera in tow. On the way, I came by some seaweed fields and discovered a sandbank in the middle, where I saw a 4-meter-long shark. I immediately panicked, turned around, and tried to capture the last moments of my life with my camera, wildly taking pictures around me, not aiming for anything. I just shouted, "Shark! Shark!" to my friend, and we both quickly got out of the water. Later, it turned out to be a nurse shark, described as sluggish, sociable, bottom-dwelling sharks that feed on invertebrates, thus posing no threat to humans. Well, now that I know what a nurse shark looks like, next time, I won't panic; instead, I'll aim for the shark and calmly capture the moment.



