
This week’s extensive special feature covers select talks from the AACR Tumor Immunology and Immunotherapy Meeting 2024 in Boston. We have organized the content by topics below.
Keynote lectures
Susan Kaech
Ton Schumacher
Biomarkers
Cornelis JM Melief
Neoantigen vaccines
Vinod Balachandran
Pablo Guasp
Lorenzo De Marco
Robert D Schreiber
Tumor immune environment
Sam Nutt
Thomas Gajewski
Roberta Zappasodi
Adjuvants
Arthur Kreig
Polina Weitzenfeld
Keynote lectures
Keynote Metabolic adaptation of T cells in cancer- Susan M. Kaech - Salk Institute for Biological Studies, La Jolla, California
In the Opening Keynote Lecture, Susan Kaech focused on the growing field of immunometabolism, particularly, how immune cells adapt to previously unseen tissues to become long-term residents, how cancer cells dysregulate the local tissue metabolic environment, and the complex interconnections that develop as changes fuel responses. Kaech’s focus for her talk was on lung and liver cancer, examining how nutrient availability and metabolic byproducts affect immune cell differentiation and function, including both suppressive and supportive effects. Analysis of tumor interstitial fluid (TIF; “tumor juice”) in two murine tumor models (compared to serum) revealed multiple classes of nutrients and metabolites that were distinct, particularly fatty acids (such as arachidonic acid) and oxidized phospholipids. As an example of the complex metabolic interplay, the use of an antibody to E06, a representative oxidized phospholipid found on dying cells, revealed a clear enrichment in the tumor compared to adjacent normal cells, with a compensatory upregulation of the scavenger receptor CD36 on infiltrating T cells, leading to increases in intracellular peroxidized lipids and potentially enhanced ferroptosis, resulting in T cell destruction. Both primary and secondary bile acids, major metabolites from the liver, also showed tumor-specific dysregulation, resulting in increases in multiple bile acids. In an SV40 T antigen-based liver cancer model, antigen-specific T cells showed an increase in bile acid uptake associated with increased exhaustion signatures (effects not seen with infiltrated T cells specific for an unrelated virus). These effects were traced to decreased viability, increased intracellular ROS, and decreased mitochondrial oxidative phosphorylation, ultimately due to slower efflux of bile acids due to lowered levels of the transporter SLC03A1. Knockout of selective bile acid synthesis pathway enzymes in tumor cells, such as BAAT, which is the primary enzyme producing conjugated bile acids, enhanced T cell infiltration, T cell function, and tumor control. All these beneficial effects were dependent on MHC presentation. Correspondingly, in human tumors, enhanced BAAT levels correlated with low CD8+ T cell levels. Secondary bile acids arising from the microbiome also impact T cells. While the bile acid LCA is inhibitory to T cell function, UDCA enhances T cell IFNγ and TNF production. In the last part of her talk, Kaech focused on how the nutrient and metabolic environment directly impact T cell phenotypes, particularly through epigenetic modifications. Histone acetylation is a key epigenetic modification, and acetyl-CoA is the primary donor for these acetyl groups, so Kaech examined the pathways leading to Acetyl-CoA production. Unexpectedly, specific acetylation events were dictated not by overall Acetyl-CoA levels, but by the nutrient source (glucose vs. acetate) driving Acetyl-CoA production, which could ultimately be traced to specific enzymes (ACSS2 or ACLY) most active in particular T cells under specific conditions. ACLY was most active in progenitor cells, with acetate as a primary carbon source, leading to maintenance of the key genes involved in the progenitor phenotype (such as Tcf), while ACSS2 was most active with glucose as a carbon source, and resulted in the upregulation of key immune checkpoints (Pdcd1 and Havcr2). Kaech was further able to show nuclear co-localization of specific acetyl transferases with one or the other of the key enzymes involved in nutrient selectivity (ACSS2 or ACLY), uncovering an unexpected locus-specific mechanism of action. Overexpression of ACSS2 enhanced tumor control in an adoptive T cell transfer mouse model. Kaech closed by suggesting that nutrient availability is a “Signal 4” for T cells, and that spatial immuno-metabolomics may begin to reveal key metabolic systems correlating with suppressive or supportive T cell features, allowing for new approaches to beneficial T cell manipulation.
Keynote Lecture: Large-scale mapping of TCR-pMHC interactions- Ton Schumacher - Netherlands Cancer Institute, Amsterdam, Netherlands
In the second Keynote Address of the conference, Ton Schumacher summarized a recent key advance in immunotherapy – neoadjuvant therapy with immune checkpoint blockade (ICB) – and a long-term aspirational goal – a sequence-level predictor for peptide:MHC–TCR interactions. Based on the major impact of ICB on advanced cancers, Schumacher and his collaborator Christian Blank considered whether and how to move ICB to earlier stages of disease. Their rationale was to treat patients with ICB with curative intent prior to surgery, at a time when neoantigens, which are required for ICB-induced T cell activation, were still present. A key concern for neoadjuvant therapy, however, was whether delay in surgery or toxicities induced by ICB might have an impact on patient outcomes; this concern was not observed in any of the following studies. An initial small biomarker study (OpACIN) of neoadjuvant treatment demonstrated a stronger T cell induction than observed with adjuvant therapy, as predicted, and encouragingly demonstrated a high frequency of pathological complete (pCR) or near-complete (pnCR) responses at surgery (6 weeks after treatment). These results were confirmed in a large study in stage III melanoma, which convincingly linked these pathological effects to enhanced long-term survival. Moving to colorectal cancer, where neoadjuvant chemotherapy had only a minor effect, even on defective mismatch repair (dMMR) CRC, the NICHE-1 study in dMMR and proficient-MMR (pMMR) with neoadjuvant ICB was conducted. Astoundingly, dMMR patients had a 100% pathological response (pR) rate (with 60% pCR) and pMMR patients had a 27% pR rate. A follow-up study (NICHE-2) in dMMR patients confirmed the high response rates and produced a 100% 3-year disease-free survival rate (median follow-up of 36.6 months). ctDNA analysis demonstrated that almost all patients had observable ctDNA prior to treatment, but 83% of patients with histological signals of tumor clearance by neoadjuvant treatment showed clearance of their ctDNA prior to surgery, suggesting a possible biomarker for reducing surgical intervention. The second part of Schumacher’s talk addressed whether the involvement of neoantigens in the immune control of tumors could be dramatically improved by “breaking the code” of neoantigen peptide:MHC–TCR interactions, thereby accelerating the application of TCR-T cells, bispecifics, and cancer vaccines. Although the magnitude of the problem is overwhelming, advances in large language models, structural modeling, and a clearer recognition of existing intellectual hurdles could be helpful. Key intellectual hurdles and solutions include (1) moving from the myopic view of TCR specificity to a single peptide to recognizing and learning from the broader cross-reactivity; (2) not limiting our exploration to naturally occurring TCRs, but instead being open to cycles of designing, testing, and re-designing synthetic TCRs that can allow faster learning of necessary rules; and (3) moving beyond the sparse data collected from patient samples to building synthetic systems that allow screening of large libraries of TCRs against large libraries of peptide:MHCs (pMHC). One such synthetic system that can be exploited is based on the previously published and now automated HANSolo approach to identify epitopes in patients that are reactive to patient T cells in a dropout screening, functional readout approach. Flipped, this approach can also be used to match TCRs in a TCR library to one or more epitopes, and large language modeling can be used to predict relevant TCR sequences for library construction that can be used to screen epitope libraries to more rapidly learn relevant rules. Combining both TCR libraries and epitope libraries (“Pair Scan”) takes this approach to a new level. Identification of interacting TCR:pMHC pairs capitalizes on enhanced adhesion due to productive TCR–pMHC pairing, separate colors for the TCR library cells and the epitope library cells, and possibly using calcium influx as a further marker of productive pairing. As an example, Schumacher showed that screening 100 expanded TCRs from TILs of a patient against 100 predicted epitopes of the patient led to the identification of 10 pairs that were statistically enriched in abundance (compared to predicted), and all 10 of these were validated following deconvolution. Duplicate Pair Screens of 40,000 members revealed comparable P values for the same pair in both screens, suggesting that recovery was correlated with avidity, possibly functional avidity, which was confirmed using killing assays with a series of 7 TCRs recognizing the same epitope.
Biomarkers
Clinical benefit from anti-HPV therapeutic vaccine and anti-PD-1 depends on high pre-treatment PD-L1 score- Cornelis Melief - ISA Pharmaceuticals, Oegstgeest, Netherlands
Therapeutic cancer vaccines involve delivering tumor antigens or neoantigens to dendritic cells, which in turn induce both CD4+ and CD8+ T cells responses. T cells can then traffick to and infiltrate the tumor microenvironment (TME), but are often hindered by tumor defenses like PD-L1, which limits T cell functionality. In recent work, Kees Melief and colleagues tested ISA101b – a synthetic long-peptide vaccine targeting an array of overlapping epitopes derived from the HPV antigens E6 and E7. This vaccine was divided into two drugs to be injected at different sites to avoid peptide competition in the vaccine-draining lymph nodes. In two clinical studies, ISA101 monotherapy was delivered s.c. in limbs of patients with HPV16+ premalignant vulvar intraepithelial neoplasia 3 (VIN3). ISA101 showed clinical efficacy; clinical responses correlated with vaccine-induced T cell responses, CR rates remained stable at 12 and 24 months, and complete histological responses were associated with viral clearance. However, to overcome later-stage disease, which has a more hostile TME, ISA101 must be combined with other interventions. In a trial of ISA101 + chemotherapy in patients with recurrent/metastatic cervical cancer, the magnitude of ISA101-induced T cell responses was associated with longer survival. In a single-arm trial of ISA101 + nivolumab (anti-PD-1) in patients with HPV+ oropharyngeal cancer (OPC), treatment had an ORR of 36%, compared to an expected 16% with nivolumab alone, and induced several long-term complete responses (40+ months). In a randomized phase II trial of ISA101b + cemiplimab (anti-PD-1) in patients with HPV+ OPC, results for the full analysis set (FAS; patients who received at least one dose of ISA101b or placebo) the ORR at 6 months showed that vaccinated patients appeared to fare worse than those treated with the placebo. However, analysis of patients who were treated with the complete protocol (3 doses of ISA101b or placebo, confirmed HPV+, and at least 1 post-baseline scan) showed a clear benefit from the addition of ISA101b. Similarly, patients who had higher PD-L1 scores (CPS ≥20) showed a clear benefit from ISA101b, with patients who had both high PD-L1 and who were treated with the full protocol showing the best ORR rates. Looking at patients’ ECOG status, a measure of how much a person’s disease affects their ability to function, Melief noted that patients who were sicker at the time of enrollment often were unable to get the full protocol treatment and were unlikely to respond, explaining the poor response rates for the full treatment population. Another study in patients with head and neck cancer who were progressive on anti-PD-1 showed evidence of durable partial responses in a minority of patients, and good survival outcomes. This data has set the stage for a phase III study in patients with PD-L1hi HPV+ cancer.
Neoantigen vaccines
Personalized RNA vaccines for pancreatic cancer- Vinod Balachandran - Memorial Sloan Kettering Cancer Center, New York, New York
Developing good cancer vaccines that activate, strong, specific, functional, and durable responses involves optimizing the antigens and delivery method, as well as considering the patient, disease, and cells involved in the immune response. While pancreatic ductal adenocarcinoma (PDAC) is an immune-cold tumor type that is not typically considered to be suited for vaccination due to low numbers of mutations and neoantigens, recent work has shown that in rare PDAC survivors, neoantigens arising from passenger mutations were targeted by endogenous T cells. Based on these findings, Vinod Balachandran and colleagues developed a novel mRNA vaccine targeting passenger mutation-derived neoantigens. In a clinical trial, personalized mRNA neoantigen vaccines containing up to 20 class I/II neoantigens were administered as an adjuvant therapy (8 priming doses and one late boost) following complete surgical resection of tumors. Patients were also treated with a single dose of anti-PD-L1 prior to vaccination and with chemotherapy after priming. T cell responses to the individual neoantigens in each patient were measured by ex vivo IFNγ ELISPOT and were confirmed using CloneTrack (a tool quantifying TCR expansion based on Vβ sequencing), together identifying high-magnitude T cell responses (up to 10% of total blood T cells) in 50% of treated patients. Detailed analysis revealed that all responding patients had expanded clones that were vaccine-specific and primarily CD8+. Further, immunological responses were correlated with delayed tumor recurrence. While these results were recently published, follow-up research is ongoing to better understand the origin of these T cell responses and whether they persist long-term. To this end, Balachandran and colleagues have evaluated peripheral blood, tumors, tdLNs, and tissue samples taken starting prior to and for up to 3.6 years after treatment, allowing for tracking of all 79 vaccine-induced T cell clones across the 8 vaccine responders. Of the 79 vaccine-induced clones, only 2 were detectable in the blood prior to vaccination, suggesting the induction of de novo responses rather than re-expansion of existing clones. After vaccination, clonal tracking allowed the researchers to estimate the trajectories of individual clones and predict how long they would persist at detectable levels in the blood after both the prime and boost. While the median predicted lifespans of neoantigen-specific T cell clones after priming was 1.1 years, boosting extended the median lifespan of clones to 7.7 years, with about 20% of clones predicted to have a lifespan of over 10 years. 70% of responding patients had clones expected to outlive their PDAC recurrence window, while 60% had clones that were expected to outlive the patient. Observational results have thus far shown roughly 80% of clones persisting 2 years or longer, into the late memory phase. This long-term persistence appeared to be a feature of vaccine-induced T cell clones, which were confirmed with cloning and neoepitope mapping by Pablo Guasp. Further, PBMCs purified up to 4 years after vaccination remained polyfunctional and cytolytic after ex vivo expansion, with both stronger and more weakly neoantigen-responsive T cells identified in 6 out of the 8 responding patients. Two patients who generated strong vaccine responses and did not recur in the 1.5 year follow-up did recur during the extended follow-up. In these two patients, the loss of function appeared to be associated with a loss of cells in the peripheral blood. One of these patients showed the shortest clonal lifespan of neoantigen-induced clones following vaccination, while the other showed the latest onset of neoantigen-induced clones following vaccination. In the patient with the late onset, neoantigen-specific clones were maintained at the time of tumor recurrence, and 7 out of 8 were found to be expanded within the recurrent tumor. Further, in another patient who showed evidence of possible PDAC recurrence shortly after vaccination, the researchers identified a lesion of dense lymphoid aggregates in the liver containing all 15 vaccine-induced T cell clones. This lesion eventually disappeared, suggesting that vaccine-induced clones may have contributed to the detection and elimination of a preclinical metastatic PDAC recurrence.
Personalized RNA vaccines induce lasting functional CD8+ T cells in pancreatic cancer- Pablo Guasp - Memorial Sloan Kettering Cancer Center, New York, New York
Building on the research presented by Vinod Balachandran on the use of personalized mRNA vaccines to prevent PDAC recurrence, Pablo Guasp presented research on how he has begun to characterize the 79 vaccine-induced T cell clones identified across the 8 responders in the phase I clinical trial. Thus far, Guasp has been able to confirm the neoantigen specificity of at least 37% of the vaccine-induced T cell clonotypes identified initially by ELISPOT and CloneTrack by stimulating PBMCs with peptides, sorting out activated cells, and performing TCR sequencing followed by TCR cloning into donor T cells. In functional analyses of clonal TCRs in donor T cells, Guasp found that many of these TCRs actually had very high avidities, despite the fact that PDAC has very few mutations and is generally not expected to contain neoantigens strong enough to elicit effective T cell responses. Looking closer at T cell responses, Guasp showed that while T cell responses to vaccine neoantigens were not evident prior to treatment, they became functionally responsive after vaccination, and remained capable of cytokine and lytic function for over 3 years in 6 out of the 8 responders. ScRNAseq and TCRseq in 6 responder patients showed that vaccine-induced clones overlapped mainly with CD8+ T cells, and that while the earliest phenotypes were mostly proliferative, they later transitioned towards effector and eventually resident-memory phenotypes. Looking at incidents of disease recurrence in patients with immunological responses, Guasp discussed the tumor cell longitudinal dynamics in two patients: one who recurred, and one who showed evidence of immune-mediated elimination of a preclinical PDAC metastasis. Based on tumor cell sequencing prior to treatment and upon tumor recurrence, Guasp identified tumor cell clones that either expanded or contracted following treatment, and found that clones that contained immunogenic vaccine neoantigens consistently contracted, indicating neoantigen-specific pruning of immunogenic cancer clones by vaccine-induced T cells.
Nous-209 off-the-shelf vaccine targets neoantigens mutations well represented both in primary and metachronous incident CRCs from Lynch syndrome patients- Lorenzo De Marco - Nouscom Srl, Rome, Italy
Cancer vaccines show promise for use in both the prevention and treatment of cancer. In order to develop an off-the shelf vaccine, Lorenzo de Marco and colleagues recently developed NOUS-209 – an off-the shelf cancer vaccine that utilizes a Gorilla Adenovirus (GAd) prime and modified Vaccinia Ankara (MVA) boost to deliver frame-shift peptides (FSPs) that are commonly shared among patients with MSI-high tumors, and are expected to be tumor-exclusive and immunogenic. To identify peptides for inclusion in NOUS-209, De Marco identified mutated mononucleotide repeats (MNR) from MSI-high tumor samples from colorectal, gastric, and endometrial cancer patients in TCGA data. This list was then narrowed down to 1087 FSPs that were present in over 5% of tumors, were absent in normal tissues, and occurred in expressed genes. From there, 209 FSP were selected to maximize patient coverage with the minimal number of genes, and these FSPs were assembled into 4 artificial genes to be carried in the GAd prime and MVA boost delivery vectors. In order to test NOUS-209, De Marco and team enrolled 45 patients with Lynch Syndrome (a genetic condition in which inherited germline mutations in mismatch-repair genes increase the risks for certain MSI-high tumors) in a Phase Ib/II clinical trial and found that after vaccination, ex vivo ELISPOT IFNγ responses to vaccine antigens could be identified in PBMCS from 100% of patients. De Marco also evaluated 38 primary tumors (identified during routine surveillance) and 20 metachronous (independent tumors identified at least 6 months after the primary tumor) and found that mutations in MNR regions in primary tumors showed considerable overlap with MNR regions in metachronous tumors. Further, frameshift mutations that were targeted with NOUS-209 were 96.1% shared, suggesting that the peptides targeted with NOUS-209 would likely capture both tumors.
Tr1 as major inhibitors of anti-tumor responses-bad help- Robert D. Schreiber - Washington University School of Medicine in St. Louis, St. Louis, Missouri
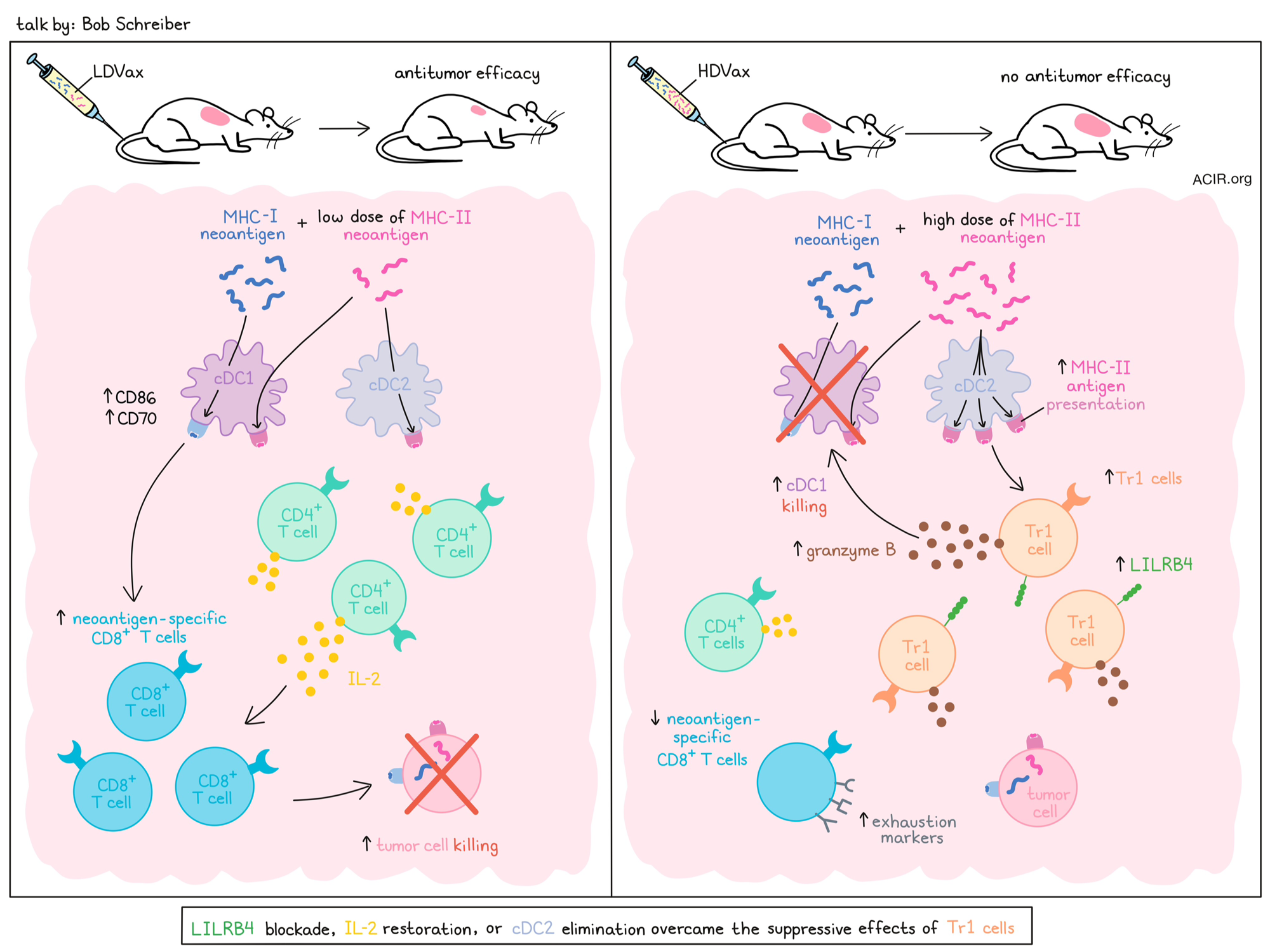
Building on his pioneering work of dissecting the MHC class I neoantigen response in the canonical methylcholanthrene-induced tumor line (129S6), Bob Schreiber described some fascinating new biology revealed by a deeper study of the dominant MHC class II neoantigen (N710Y-mItbg1) in these cells. In a model system with low neoantigen burden, but knocked-in class I (mLama4) and class II (mItbg-1) neoantigens, T cell responses to both neoantigens were found to be required for tumor control. While tumor-controlling prophylactic vaccination with increasing amounts of the class I neoantigen mLama4 with a fixed low dose of the class II neoantigen mItbg-1 identified a threshold dose at or above which tumors were controlled, fixing the class I neoantigen dose (at the effective threshold level), while varying the class II neoantigen dose required only a very low dose of the class II neoantigen to be effective (Low Dose Vax; LDVax). Surprisingly though, tumor control was lost at higher doses (High Dose Vax; HDVax). In adoptive transfer studies, transfer of only a 1/25 ratio of HDVax/LDVax T cells inhibited tumor rejection by LDVax cells. Phenotypic and single-cell sequencing indicated that HDVax-induced suppressive cells were not Tregs, but had properties of previously described Tr1 suppressive cells, marked by the surface marker LILR4B, with or without Semaphorin 4A. Gene set enrichment analysis confirmed the cytotoxic Tr1-like expression pattern, which marks a suppressive cell type associated with tolerance, autoimmune disease, and some chronic viral infections, but developmentally distinct from Foxp3+ Tregs. Genetic knockouts indicated these cells were induced by cDC2s, and were cytotoxic to cDC1s, both in vitro and in vivo. Cytotoxicity was dependent on granzyme B, and suppression could be inhibited with an antibody to LILRB4. IL-2 expression was low in these cells, so given the important role of IL-2 in CD8+ T cell differentiation, IL-2 supplementation was evaluated to overcome the reduced production. A CD8-targeted, cis-acting IL-2 (being developed by AsherBio) was tested in vivo and was shown to overcome the effect of inhibitory Tr1-like cell formation by HDVax, leading to increased neoantigen-specific CD8+ T cell frequency; wild-type IL-2 was significantly less effective. To determine if the generation of Tr1 cells was uniquely related to a peptide vaccine or was a natural phenomenon correlating with tumor growth, Schreiber examined the level of Tr1 cells in tumors treated with anti-PD-1, beginning shortly after tumor implantation (still sensitive to anti-PD-1) or at later resistant timepoints. Tr1 cell accumulation in tumors coincided with the onset of anti-PD-1 resistance, while Treg cell numbers remained constant, indicating the effect was not unique to peptide vaccination. A key future development will be the identification of the LILRB family member (a larger gene family) in humans, which is critical.
Tumor immune environment
Competition for dendritic cells limits TCR-T cell activation in tumor-draining lymph nodes and impairs synergy with PD-L1 blockade - Sam Nutt - Fred Hutchinson Cancer Center, Seattle, Washington
Adoptive cell therapy (ACT) and immune checkpoint blockade (ICB) are both highly effective immunotherapies, but the combination of the two hasn’t always synergized as well as might be expected based on their mechanisms of action. Looking for clues as to why this might be, Sam Nutt turned to the tumor-draining lymph nodes, which play a key role in promoting antitumor responses. Under normal priming conditions, progenitor exhausted PD-1+Tcf1+ T cells (Tpex) slowly generate a population of T cells that go on to infiltrate tumors, but with adoptive cell transfer, a high volume of T cells is introduced at once. Nutt hypothesized that this might force T cells into competition for interactions with a limited supply of DCs, which are reduced during lymphodepletion. Using an OVA-expressing KP tumor model, Nutt compared endogenous antigen-specific T cell responses and TCR-T cell responses, and found that in both settings, T cells preferentially accumulated in tdLNs versus non-tdLNs. However, while endogenous SIINFEKL-specific T cells uniformly upregulated PD-1, TCR-T cells did not, with a population of TCR-T cells remaining PD-1-. Further, TCR-T cells that did express PD-1 expressed it at lower levels, suggesting weaker activation. Imaging of tdLNs showed that while endogenous T cells were distributed throughout the lymph nodes, both near and far from DCs, TCR-T cells formed large clusters around DCs, indicating competition. To test whether this competition was limiting optimal DC engagement and T cell activation, Nutt used a KP-OVA tumor model treated with cyclophosphamide and different doses of TCR-T cells. While cyclophosphamide reduced DCs, lowering the TCR-T cell dose reduced the T cell:DC ratio, which, in turn, correlated with increased PD-1 expression. Looking at other markers of activation and functionality, Nutt found that compared to endogenous Tpex cells, TCR-T Tpex cells produced less IL-2 and TNFɑ. When these Tpex subsets were transferred into mice with implanted KPOVA cells to test T cell memory recall responses, endogenous T cells outperformed TCR-T cells in terms of tumor control. Finally, Nutt has begun to evaluate models of ACT in combination with checkpoint blockade, and has found that low doses of transferred TCR-T cells are able to mediate the same amount of tumor regression as doses 100x higher, suggesting that reducing the competition in the tdLN may promote responsiveness to anti-PD-L1. Moving forward, Nutt intends to evaluate how competition affects T cell differentiation states, and whether these findings will translate to additional models.
Spatial transcriptomics and novel insights into the immune tumor microenvironment- Thomas Gajewski - University of Chicago, Chicago, Illinois
Thomas Gajewski highlighted the intricate dynamics among immune cells – particularly dendritic cells (DCs) and CD8+ T cells within the tumor microenvironment – and the critical role of spatial transcriptomics in unraveling interactions that occur during immunotherapy. He emphasized two distinct categories of tumors: inflamed and non-inflamed, with the former being characterized by the presence of immune cells, making them more responsive to therapies such as checkpoint inhibitors, while the latter remains resistant due to the lack of immune cell infiltration. A key component of inflamed tumors is the dendritic cell-produced chemokines and cytokines, which recruit T cells into the tumor. Earlier work from Gajewski and colleagues found that tumor cell-intrinsic β-catenin activation disrupts the recruitment of dendritic cells and T cell priming, ultimately leading to non-inflamed TMEs and resistance to checkpoint blockade and adoptive T cell therapies. Further studies revealed that β-catenin-driven tumors lack a specific subset of dendritic cells known as Batf3-lineage DCs, which are necessary for both the recruitment and the activation of CD8+ T cells within the tumor site. Notably, the depletion of dendritic cells in mouse models prior to PD-1 blockade therapy also resulted in a loss of therapeutic efficacy, underscoring the importance of dendritic cells in sustaining antitumor T cell responses. To determine whether Batf3-lineage DCs are required at the effector phase of the antitumor immune response at the tumor site, the researchers generated a genetically engineered mouse model to deplete Batf3-lineage DCs. Dendritic cell depletion in mouse models before PD-1 blockade resulted in a loss of therapeutic efficacy and CD8+ T cell expansion, underscoring the importance of dendritic cells in sustaining antitumor T cell responses. Human data further supported these findings. Multiplex imaging and spatial transcriptomics revealed a strong association between CD8+ T cells and dendritic cells within the TME. This interaction was predictive of better outcomes in patients with melanoma treated with anti-PD-1 therapy, as spatial proximity between the two cell types correlated with a transcriptional profile indicative of active T cell responses. These results suggested that the presence of CD8+ T cells alone was not sufficient for effective immunotherapy, but rather their functional interaction with dendritic cells was crucial for driving durable antitumor immunity. These results were consistent with prior studies showing that in vitro Flt3 ligand-induced and -activated DCs injected intratumorally could restore immune infiltration and sensitize β-catenin-driven mouse tumors to checkpoint inhibitors. To demonstrate whether the immunosuppressive role of β-catenin could be reversed, the researchers genetically engineered a mouse model with doxycycline-inducible β-catenin. As anticipated, doxycycline-induced β-catenin tumors showed decreased T cell infiltration and blunted the efficacy of anti-PD-L1 and anti-CTLA-4 therapy. Removal of doxycycline resulted in β-catenin loss and re-established T cell infiltration within the TME. However, β-catenin removal from an established tumor did not restore dual checkpoint blockade efficacy. scRNAseq revealed an altered macrophage transcriptional program in β-catenin-removed tumors, with the expansion of suppressive/M2-like gene expression patterns. Spatial transcriptomics further revealed expanded M2-like macrophages interacting with T cells, which lacked the activation signature observed for T cells near DCs in the TME, suggesting that β-catenin alters the tumor microenvironment unfavorably, with persistent M2-like cells at the early stages of tumor development. In summary, Gajewski highlighted the importance of spatial transcriptomics as a powerful tool for understanding the complex cellular interactions within the TME. His findings emphasized the need for continued exploration into dendritic cell biology, β-catenin signaling, and myeloid cell reprogramming to overcome the barriers to effective immunotherapy in resistant tumors.
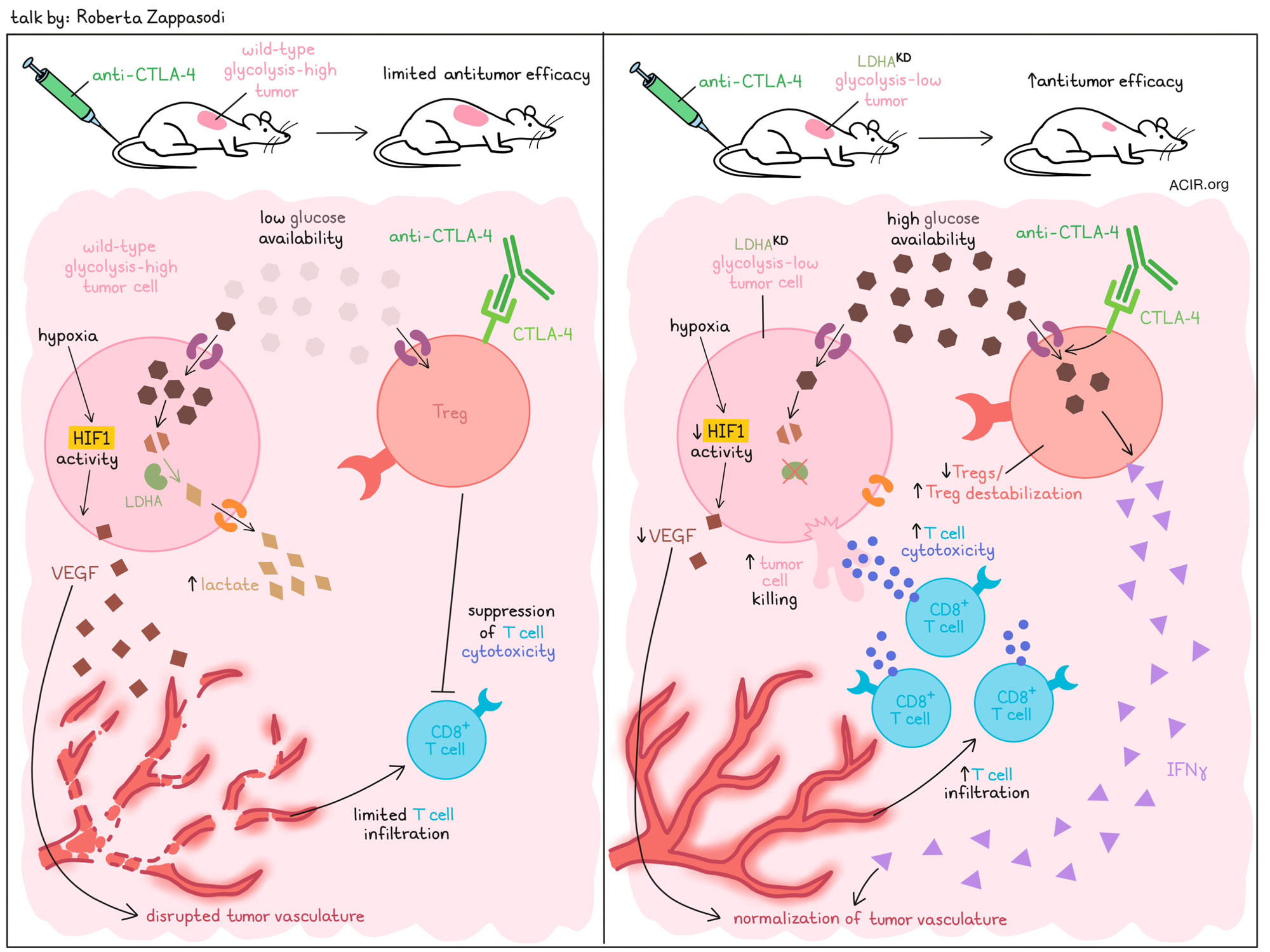
Rational immunotherapy approaches based on the tumor glycolytic state- Roberta Zappasodi - Weill Cornell Medicine, New York, New York
Immune checkpoint blockade is highly successful in a subset of patients, but not all patients respond, in part due to immune resistance mechanisms in the tumor microenvironment (TME). Investigating such mechanisms, Roberta Zappasodi described how tumors often increase glycolysis, resulting in TMEs marked by low glucose availability and increased lactate. Across several tumor types, features of increased glycolysis, including high expression of LDHA (an enzyme involved in the conversion of pyruvate to lactate during glycolysis), were associated with reduced T cell infiltration, reduced cytotoxicity, and worse patient survival outcomes. To test whether reducing glycolysis could increase T cell infiltration and immunotherapy responses, Zappasodi generated tumor models with reduced glucose uptake and glycolytic capacity through knockdown of LDHA (LDHAKD). Mice bearing LDHAKD 4T1 tumors showed improved responses to treatment with anti-CTLA-4 (but not anti-PD-1). Interestingly, these LDHAKD tumors were marked by loss of cells expressing the Treg markers CTLA-4 and CD25. Following the hypothesis that Tregs were feeding on the increased glucose available in LDHAKD tumors, Zappasodi found that when cultured Tregs were treated with anti-CTLA-4, they increased glucose uptake and produced more IFNγ, indicating Treg destabilization. Further, anti-CTLA-4 reduced the capacity of Tregs to suppress CD8+ T cells, but only when glucose levels were high. To model this in vivo, researchers used mouse models in which Glut1 (glucose uptake) was knocked out specifically in Tregs. In this setting, Tregs were not depleted by anti-CTLA-4 in LDHAKD tumors (now using B16 to accommodate the C57BL/6 mouse model), confirming the role of increased glucose uptake by Tregs in reducing their numbers in such tumors. These results suggest that anti-CTLA-4 may be most suited for the treatment of glycolysis-low (glucose-high) tumors. In more recent research, Zappasodi’s lab has begun to investigate how IFNγ-producing Tregs in the tumor contribute to antitumor immunity. Based on previous studies showing that ICB-induced increases in IFNγ production by CD4+ T cells normalize tumor vasculature, Zappasodi and colleagues evaluated the vasculature in the wild-type and LDHAKD 4T1 tumors. They found that in the glycolysis-low LDHAKD tumors, anti-CTLA-4 increased signatures for high endothelial venules and stabilizing pericytes, and decreased an angiogenic pericyte signature, suggesting an overall more desirable vasculature. Further, LDHAKD tumors showed reduced responses to hypoxia (HIF-1 activity), leading to reduced VEGF secretion (neo-angiogenesis), and more normalized tumor vasculature with less leakiness. At the cellular level, the researchers identified reduced CD31+ endothelial cells and NG2+ pericytes (associated with pathological conditions), and increased PDGFRβ+ pericytes (associated with blood vessel stabilization) in LDHAKD tumors. These features were associated with increased infiltration of CD8+ T cells, effector T cells, and Tregs in the tumors, suggesting that overall, reducing tumor glycolysis enhances T cell tumor infiltration. In order to improve the effects of anti-CTLA-4 in glycolysis-high tumors, Zappasodi’s team investigated a combination therapy with a VEGF inhibitor. Alone, anti-VEGFR2 slightly improved responses in glycolysis-high, but not in glycolysis-low tumors (likely due to the more disrupted vasculature in the glycolysis-high setting). In combination with anti-CTLA-4, anti-VEGFR2 induced long-term survival, associated with a decrease in tumor endothelial cells (including those expressing VEGFR2) and reduced vasculature leakiness. Finally, evaluating the clinical relevance of these findings, Zappasodi showed that human TNBC shows disorganized vasculature, and that endothelial cell interactions via VEGF are lower in human LDHA-low TNBC. Further, TCGA data showed positive correlations between tumor glycolysis and an angiogenic phenotype, suggesting that tumor glycolysis may serve as a biomarker to select patients for vasculature-normalizing therapy in combination with immunotherapy. Another strategy to treat glycolysis-high tumors could be to deplete Tregs. Investigating this, researchers found that Treg-depleting anti-CTLA-4 therapy had stronger effects in glycolysis-high tumors, both in terms of initial tumor clearance and response to rechallenge. Evaluating the difference in memory responses, they found that in glycolysis-low tumors, anti-CTLA-4 was likely less effective due to the depletion of destabilized IFNγ+ Tregs, which are beneficial in the TME. In human tumor samples, Tregs were increased and correlated with tumor glycolysis, suggesting that high glycolysis may also be a potential biomarker to select for patients who could benefit from Treg depletion.
Adjuvants
Retroviral mimics costimulate TLR7/8/9 for enhanced activation of dendritic cells and myeloid cells to induce anti-tumor CD8+ T cells- Arthur Krieg - University of Massachusetts Chan Medical School, Needham, Massachusetts
Arthur Krieg delved into the therapeutic potential of targeting nucleic acid-sensing pathways, specifically focusing on the nucleic acid-sensing toll-like receptors (TLRs) 7, 8, and 9, which have evolved as the signals used by innate immunity to eliminate virally and retrovirally infected cells. TLR7/8 recognizes uridine-rich viral RNA, while TLR9 detects unmethylated CpG DNA motifs. Although treatment with the TLR9 agonist vidutolimod has shown promise in anti-PD-1-refractory melanoma, inducing CD8+ T cells and systemic tumor regression in a subset of patients, Krieg emphasized that engaging TLR9 with DNA alone is insufficient. Activation of all three TLRs is required to induce a better endogenous antitumor CD8+ T cell response, and important attention must be paid to which specific TLRs are being targeted, which cell types are being affected, and which distinct type I interferon responses are being induced. Krieg began by discussing the importance of selectively inducing IFN-α (compared to IFN-β) in human cancer immunotherapy. IFN-α drives stronger antitumor CD8+ T cell responses, whereas IFN-β tends to induce regulatory T cells and IL-10 production. Only IFN-α has been approved for anticancer or antiviral therapy. Plasmacytoid dendritic cells (pDCs) are the dominant producers of IFN-α in response to viral stimuli, and are uniquely equipped to sense viral RNA and DNA through TLR7 and TLR9. There are two subsets of pDCs: P1 pDCs (PD-L1+/CD80-) produce IFN-α while P3 pDCs (PD-L1-CD80+) promote Th2-like responses. Of all tested innate immune activators, CpG-A DNA (a TLR9 agonist) induced the highest number of P1 pDC and best mimicked the viral/retroviral DNA genomic structure and, following packaging in a virus-like particle, best associated with efficacy in refractory advanced melanoma. Krieg noted that clinical trials have shown promising results, particularly as neoadjuvant therapy and when combined with checkpoint inhibitors like anti-PD-1 or anti-CTLA-4 antibodies. These combinations have led to significant tumor regressions in patients who previously had limited responses to checkpoint inhibitors alone. Responses were associated with increased CD8+ and CD4+ TILs, pDCs, tertiary lymphoid structures, and macrophage activation signatures. He presented a case study of a 48-year-old female with heavily pre-treated advanced melanoma with a lung metastasis, who was injected with vidutolimod in a melanoma groin lesion, and who showed clearance of the metastasis and a lasting response, demonstrating a strong systemic effect. Krieg also underscored the importance of delivery methods in maximizing the efficacy of TLR9 agonists. A significant obstacle to vidutolimod’s efficacy is the rapid induction of antibodies against its virus-like particle (VLP) delivery system. High antibody titers dampen the pDC response, correlating with poor treatment outcomes. Krieg proposed strategies to overcome this limitation with a lipid nanoparticle delivery system that avoids inducing neutralizing antibodies. Furthermore, activation of TLR7/8 using GU-rich RNA with a phosphodiester backbone, along with a TLR9 agonist, mimicked the complete retroviral/viral genome and provided the greatest immune induction. Separate agonists for TLR9 and TLR7/8 also provided better control and the possibility to fine-tune the doses for male and female patients, since females show higher TLR7/8 responses. Kreig also highlighted that CD36 was associated with M2-like macrophages, and nucleic acid therapeutics may be targeted systematically to M2-like TAMs using a lipid delivery system. Intravenous delivery of a standard lipid nanoparticle containing a GU-rich phosphodiester backbone RNA as a TLR7/8 agonist and CpG-A as a TLR9 agonist activated blood myeloid cells in non-human primates. In summary, retroviral mimics that activate TLR7/8/9, particularly using CpG-A, show great potential in stimulating robust CD8+ T cell responses and systemic tumor regression.
Fc-optimized agonistic CD40 antibody induces tumor rejection and systemic antitumor immunity: From the bench to the bedside and back- Polina Weitzenfeld - The Rockefeller University, New York, New York
CD40 is a transmembrane protein of the TNFR superfamily, expressed mainly on antigen-presenting cells such as dendritic cells (DCs). Interaction with its ligand (CD40L) activates downstream signaling that leads to the activation and maturation of DCs, which is pivotal for initiating an antitumor immune response. Although therapeutic CD40-targeting antibodies have been explored in the clinic, most first-generation antibodies failed due to limited efficacy and severe toxicity, often resulting in liver damage and halting their development. To improve the therapeutic impact and reduce adverse events associated with targeting CD40, Polina Weitzenfeld and colleagues focused on two key aspects: changing the treatment administration route from systemic to intratumoral, and re-engineering the Fc portion of the antibody. Efficient activation of CD40 requires its trimerization, and the previous generation of anti-CD40 antibodies failed to trimerize CD40 successfully (CD40L is a natural trimer). The researchers enhanced the binding affinity of the Fc region of the anti-CD40 antibody for FcRIIb, which is crucial for promoting CD40 trimerization – a necessary step for its full activation. The novel Fc-engineered CD40 antibody generated via point mutation in the Fc region of the human anti-CD40 antibody (clone 2141 with variant V11) demonstrated a remarkable 96-fold higher affinity for FcRIIb and a low affinity for FcRIIa, minimizing the risk of toxicity. In an aggressive E0771 breast carcinoma model, systemic or intratumoral administration of non-engineered anti-CD40 antibodies showed no effect, but the intratumorally administered Fc-engineered antibody led to complete tumor regression in transgenic mice expressing human CD40 and FcγR. The response extended beyond local tumors, as the untreated distal lesions also regressed, indicative of a robust systemic immune response and abscopal effect. Beyond immediate tumor clearance, the antibody also induced long-term immune memory, as demonstrated by a rechallenge experiment in which treated mice rejected new tumor inoculations without additional therapy. Cytokine and chemokine profiling revealed that the antibody-induced systemic transient immune activation peaked after the second dose. The Fc-engineered anti-CD40 antibody also showed promising results in an ongoing phase 1 clinical trial, with only low-grade adverse events (grade 1-2 fever, injection site reactions) and no dose-limiting toxicities. Among 12 treated patients, the overall response rate was 20%, including complete responses in some patients with breast cancer and melanoma. One notable case was a heavily pretreated, 67-year-old patient with breast cancer and widespread metastases, who experienced complete regression of injected and non-injected tumors. Intratumoral 2141-V11 induced tumor-infiltrating lymphocyte (TIL) recruitment and tertiary lymphoid structure (TLS) formation in patients and animal models, indicating a robust immune response. Spatial analysis indicated a close alignment of B cells and DCs. In summary, Fc-engineering of CD40 antibodies can enhance antitumor efficacy, induce systemic immunity, and promote durable tumor clearance with a favorable safety profile, paving the way for its broader therapeutic use in oncology, with ongoing trials in bladder cancer, prostate cancer, and glioma. Multiple Phase II trials are underway.
Back to Top
By Lauren Hitchings, Shishir Pant, Ute Burkhardt, and Ed Fritsch



