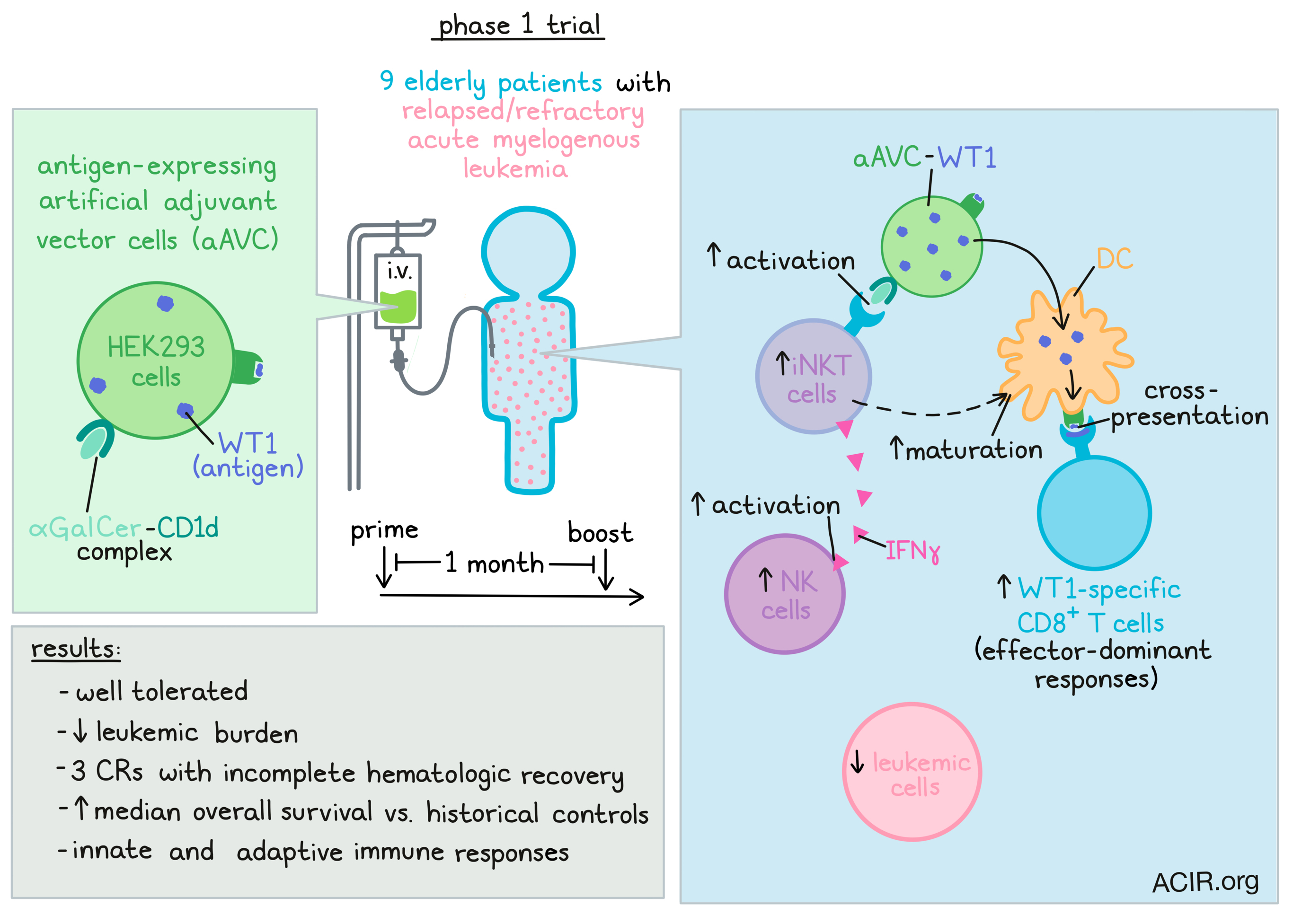
Strategies that induce robust activation of both the innate and adaptive branches of the immune system are warranted for effective immunotherapy. To this end, Fujii, Kawamata, and Shimizu et al. developed a novel therapeutic vaccine platform consisting of antigen-expressing artificial adjuvant vector cells (aAVC). This strategy was assessed in a phase I trial including nine elderly patients with relapsed/refractory acute myelogenous leukemia (AML). The efficacy and safety data were recently published in Molecular Therapy: Oncolytics.
Invariant natural killer T (iNKT) cells are CD1d-restricted innate-like αβ-T cells expressing an invariant T cell receptor (TCR). These cells can be stimulated by α-galactosylceramide (α-GalCer) bound to CD1d to induce the production of IFNγ, activation of NK cells, and in situ maturation of dendritic cells (DCs). The aAVCs used in this study were allogeneic HEK293 ectopically expressing the AML tumor antigen Wilms’ tumor antigen 1 (WT1), which was expressed intracellularly, and CD1d, which could form an αGalCer-CD1d complex on the cell surface.
Patients were intravenously treated with two cycles (prime-boost, 1-month dosing interval) of aAVC-WT1 in this phase I trial. No dose-limiting toxicities were found, and the therapy was well tolerated. The leukemic burden was used to assess the efficacy of the treatment. The number of leukemic cells decreased by over 15% in 7/9 patients, and in five patients the reduction was over 50%. Three patients with >15% reduction had a complete response with incomplete hematologic recovery (CRi). The median overall survival for these patients was 360 days, which is demonstrably better than historical data (4.5-4.9 months).
The researchers then assessed the mechanism of action of the treatment by determining the frequency of iNKT, NK, and T cells in the peripheral blood (PB) and bone marrow (BM) before and after treatment. iNKT cells and NK cells increased after treatment in the BM and/or PB in all but one patient. Treatment increased in vitro ligand-dependent IFNγ production in two patients, suggestive of iNKT cell-mediated responses. Additionally, one patient had ligand-independent IFNγ production, suggestive of activation of iNKT or NK cells. Additionally, six patients had NK cell activation and enhanced functioning of innate lymphocytes as assessed by qPCR for IFNG expression.
Four HLA-A24+ patients were analyzed for the presence of WT-1 tetramer+ CD8+ T cells using a known HLA-A24 epitope. The total number of WT1 tetramer+ CD8+ T cells increased in 3/4 patients in PB and/or BM, with the peak expression at 1-2 weeks after treatment. Since the vector cells used were HLA-A*02:01 and not HLA-A24, these could not directly present WT1 to these tetramer+ T cells, suggesting DC-mediated antigen cross-presentation had taken place in these patients.
In four patients who experienced a ≥50% reduction in leukemic cells in the BM after therapy, adaptive immunity targeting WT-1 was further assessed. PBMCs were cultured with an overlapping 15mer peptide library spanning the entire WT-1 protein. Furthermore, in two patients with the best clinical responses, ten sub-pool mixtures including 12-13 peptides each were assessed. In all four patients, there were increases in the frequencies of antigen-specific CD8+ T cells after treatment, while only low frequencies were detected prior to treatment. These T cell clones were able to produce IFNγ and/or TNFα. The frequency of these cells was low, but they were detected for prolonged periods of time (6-12 months), suggestive of memory formation. In patients with leukemic blast reductions, the most reduction was seen around the time of the peak CD8+ T cell responses.
The researchers then used scRNAseq to assess gene signatures of T cells obtained from the BM and PB of one patient who experienced long-term leukemic growth reduction. In the BM, effector markers were increased, and cytotoxicity-related molecules were expressed at higher levels in the BM than in the PB.
Next, the T cell heterogeneity in four patients who had detectable WT1-specific T cell responses was assessed. In the first patient, two effector and a pre-exhausted resident memory (Trm) CD8+ T cell cluster increased after therapy. In the second patient, effector CD8+ T cells and a pre-exhausted Trm CD8+ cluster increased, while effector and dysfunctional pre-exhausted (Tpex) CD8+ clusters, as well as regulatory T cell and follicular helper CD4+ clusters decreased. The third patient experienced a decrease in pre-exhausted Trm and IFN-related effector CD8+ clusters. Finally, the fourth patient had an increase in two FGFBP2-type effector CD8+ clusters, a Tpex CD8+ cluster, a naive CD4+ cluster, and an unclassified CD4+ cluster. On the other hand, a pre-exhausted Trm CD8+cluster, a central memory CD4+ cluster, and regulatory CD4+ clusters decreased. Overall, an effector-dominant response was seen in these patients, even though patterns of clusters varied slightly.
Next, the researchers determined whether there was an expansion of pre-existing clones or new clones were formed for these effector-dominant responses. Single-cell and bulk TCRseq was performed on cells obtained from the BM and PB of three patients. Most of the clonotypes present in these patients did not change after treatment. However, in one patient, several pre-existing clonotypes expanded after treatment, while in two patients, new clones were detected after therapy. The clonal frequencies and kinetics of CD8+ T cell clones shared between PB and BM were assessed. The clonal frequencies of total shared CD8+ T cell clones accumulated in the BM did not alter after treatment, but remained high (30-50% or 70-90%). The shared T cell clonotypes comprised transient, medium-lived, and long-lived clones.
Evaluation of the kinetics of CD8+ T cell clonotypes revealed that new clones were mostly transient (disappearing by day 42) or of a stable subtype (persisting at day 42). Increased clones had upregulation of cytotoxic genes, while exhaustion markers were reduced. New clones, emerging at day 14 or 42, expressed high levels of immune checkpoint molecules, including CTLA4, PDCD1, and TIGIT, suggestive of recent activation. Clones that decreased after therapy expressed low levels of cytotoxic markers and high levels of exhaustion markers, suggestive of terminally exhausted T cell subsets. Tracing individual clones in two patients showed that long-lasting clones were mainly derived from increased or decreased clones, while new clones were short-lived.
Collectively, this novel vaccine platform targeting a single tumor-associated antigen was safe and effective in inducing innate and long-lived adaptive antitumor responses. Importantly, the clinical outcomes were encouraging in this difficult-to-treat relapsed/refractory AML population. Further assessment in clinical studies will determine long-term efficacy in AML and other cancers.
Write-up by Maartje Wouters, image by Lauren Hitchings



