Despite exciting clinical success, resistance to immune checkpoint inhibitors remains a widespread problem, and researchers are always on the hunt for new treatment targets that might improve T cell functionality. In a recent paper published in The Journal of Clinical Investigation, Stanczak and Siddiqui et al. set out to address this problem by exploring the role that Siglecs might play in the activation of tumor-infiltrating T cells.
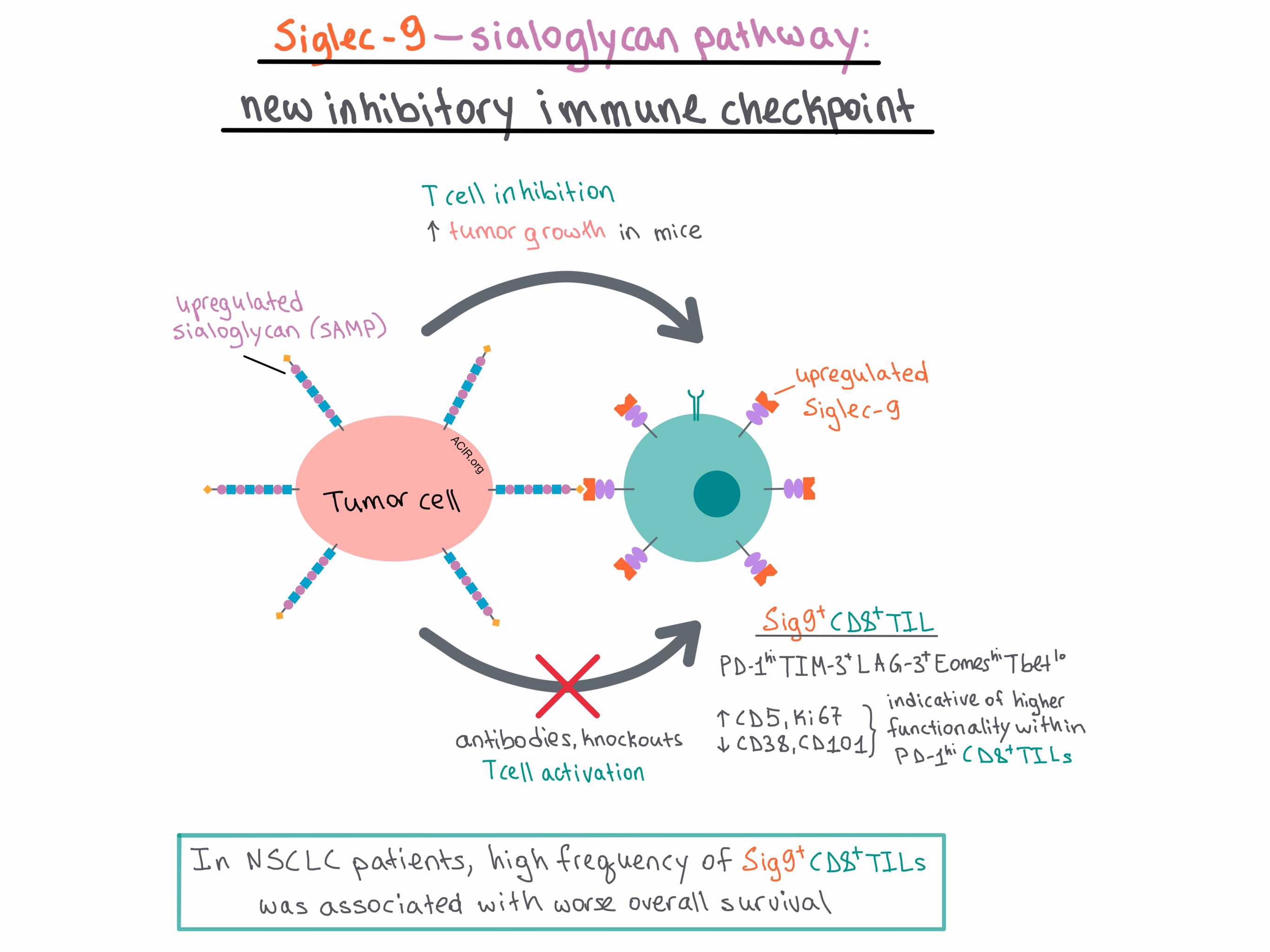
Previous research has shown that in a tumor setting, the binding of sialic acid-binding immunoglobulin-like lectin (Siglec) family members to their ligands, sialoglycans (which serve as “self-associated molecular patterns”, or SAMPs), can lead to the inhibition of natural killer (NK) cell-mediated tumor killing, induce a tumor-associated macrophage phenotype, and promote myeloid cell-mediated cancer progression, overall contributing to immune evasion. However, whether Siglecs are involved in intratumoral T cell activity has not been explored. Therefore, the researchers started out by analyzing immune cells in primary non-small-cell lung cancer (NSCLC) samples for Siglec expression. As expected, Siglecs were expressed on NK cells and myeloid cells. Although Siglec expression is very low in T cells from peripheral blood of healthy donors or non-malignant splenic tissue, several Siglecs were expressed on a significant portion of tumor-infiltrating lymphocytes (TILs), including CD4+ and CD8+ T cells. The expression of Siglec-9 on TILs was most prominent and most consistent across patients, and so the team focused the rest of their analysis on Siglec-9. Elevated levels of Siglec-9 were also observed in TILs from colorectal cancer and epithelial ovarian cancer.
Flow cytometry demonstrated that Sig9+CD8+ TILs in NSCLC were Eomeshi and Tbetlo and co-expressed other inhibitory receptors, including PD-1 (usually PD-1hi), TIM-3, and LAG-3. Sig9+CD8+ T cells in general exhibited higher upregulation and co-expression of inhibitory receptors than Sig9-CD8+ TILs in NSCLC.
When activated, Sig9+CD8+ TILs expressed significantly higher levels of activation markers (such as CD25 and CD69) compared with Sig9-CD8+ TILs. Although CD8+ TILs were dysfunctional, as indicated by reduced production of IFNγ and TNFα, interestingly, it was easier to reinvigorate the Sig9+CD8+ TILs than Sig9-CD8+ TILs. This may be because Sig9+CD8+ TILs exhibited a higher CD5 and Ki67 expression and lower CD38 and CD101 expression, a pattern that has previously been associated with higher functionality within PD-1hiCD8+ TILs. Overall, the results of Sig9+CD8+ TIL characterization suggest that these cells constitute a distinct subpopulation of tumor-specific CD8+ TILs.
To explore the functional role of Siglec-9 on T cells, the researchers utilized the high affinity Siglec ligand LGALS3BP, which is upregulated in NSCLC, and found that it inhibited CD8+ T cell activation. On the other hand, downregulation of sialoglycan-SAMPs (Sia-SAMPs) via knockdown of GNE (an enzyme involved in the production of sialic acid) on tumor cell lines led to higher T cell-mediated killing in a co-culture assay in the presence of a bispecific antibody, with the Sig9+CD8+ TILs showing higher reactivity than Sig9-CD8+ TILs. The team also found that bivalent, agonistic Siglec-9 antibodies inhibited T cell activation in a dose-dependent manner. Meanwhile, a monovalent Fab fragment that inhibited Sia-SAMP-Siglec-9 interactions by interfering with ligand binding led to increased T cell activation. Together these results suggest that the Sia-SAMP-Siglec-9 interactions can be targeted to enhance TIL activation in NSCLC.
In order to investigate Siglec-9 interactions in vivo, the team took a look at the murine equivalent (Siglec-E) and found that it was upregulated on CD8+ TILs in mice with various subcutaneous tumors when compared with splenocytes from both tumor-bearing mice and healthy controls. The phenotype of murine SigE+CD8+ TILs was similar to that of the human Sig9+CD8+ TILs. The team then compared the growth of subcutaneous wild-type (WT) and GNE-deficient MC38 colon carcinoma tumors in mice and found that the GNE-KO MC38 tumors, which had a downregulated expression of Sia-SAMPs, exhibited delayed tumor growth. This was also observed in GNE-KO EMT6 breast carcinoma tumors. The GNE-KO MC38 tumors had an elevated infiltration of classical DCs and CD3+, CD8+, and CD4+ T cells. There was no difference in infiltration of other myeloid cells or Tregs. In a separate experiment, MC38 and EMT6 GNE-KO tumor growth was shown to be similar in WT and adaptive immune system-deficient NSG mice, leading the authors to conclude that sialylation directly affected tumor growth.
In transgenic mice that selectively expressed human Siglec-9 (which is capable of binding to murine sialylated ligands) on CD4+ and CD8+ T cells, MC38 tumors grew faster and larger compared with controls. These results support the idea that inhibitory Siglecs on T cells are able to mediate tumor evasion. In a separate mouse model, where Siglec-E was engineered to switch from an inhibitory to an activating receptor, MC38 tumors grew slower and smaller, and this effect was dependent on CD4+ and CD8+ T cells. Adoptive transfer experiments further demonstrated that Siglec-E on T cells is functionally involved in immune evasion.
Finally, the authors found that NSCLC patients with a high frequency of Sig9+ TILs (>30% of CD8+ T cells) had worse overall survival, although the significance was reduced when age and stage were taken into account. In addition, they observed that certain polymorphisms of Siglec-9 were associated with reduced risk of developing squamous cell lung cancer and colorectal cancer, suggesting that Siglec-9 may play a role in the development of these cancers.
Overall, Stanczak and Siddiqui et al. show that the Sia-SAMP:Siglec-9 pathway is a newly defined inhibitory immune checkpoint for T cell activation, and targeting this pathway may improve antitumor response.
by Anna Scherer



