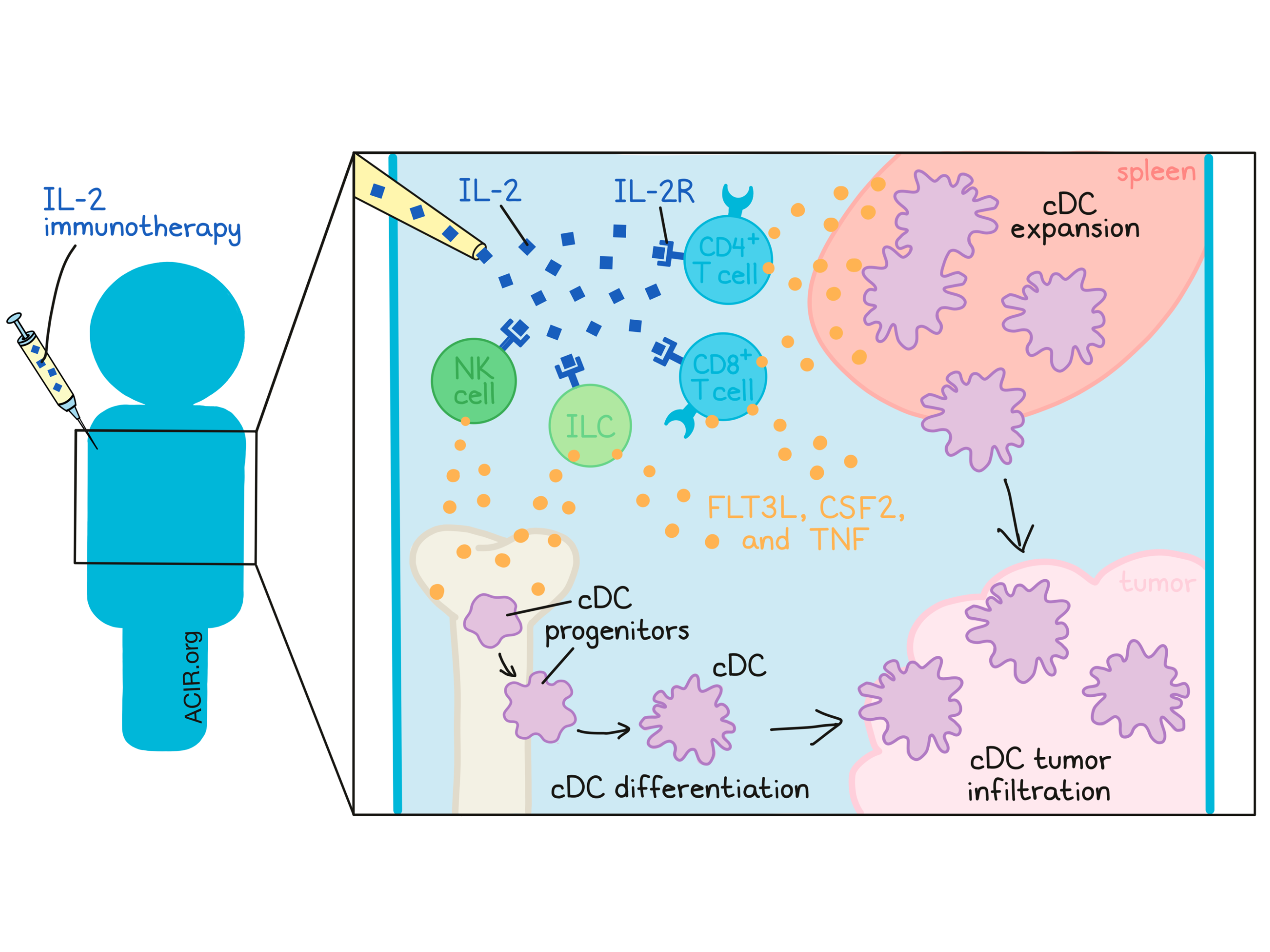
Interleukin-2 (IL-2) treatment was the first FDA-approved cancer immunotherapy, achieving robust and curative responses in metastatic melanoma and renal cell cancer in the 1980s-90s and paving the way for advances in the decades since [1]. Mechanistically, IL-2 has been understood to primarily influence T cell expansion and activation. However, the role of IL-2 in regulating activity and homeostasis of conventional dendritic cells (cDCs), essential in T cell priming, remains ambiguous. In research recently published in Science Translational Medicine, Raeber et al. report on a critical role of IL-2 immunotherapy in expanding cDCs for effective antitumor immunity.
Beginning their investigation by injecting IL-2 into wild-type mice, Raeber et al. observed increases in splenic cDC proliferation and abundance. An IL-2/anti-IL-2 antibody complex (IL-2cx, which preferentially targets the dimeric IL-2Rβγ [CD122/CD132] complex) functioned similarly, expanding cDC1s and cDC2s, but not plasmacytoid DCs in lymphoid organs. Notably, these results were mirrored in a clinical trial of recombinant IL-2 (aldesleukin). Compared to pre-therapy samples, higher Ki67 expression in blood cDC1s and cDC2s was observed after treatment, indicating increased proliferation of these cells. While consistent between mice and humans, these results were unexpected, given the unknown function of IL-2 in DC proliferation.
To investigate potential mechanisms underlying this expansion, the authors first evaluated cell cycle stages of mature cDCs. In untreated mice, ~5% of splenic cDCs were actively cycling, compared to ~16% in mice treated with IL-2cx, showing increased proliferation. Next, the researchers studied cDC progenitor populations. Interestingly, monocyte/DC progenitor, common DC progenitor, and pre-cDC2 numbers were reduced in the bone marrow, concomitant with increases in cDC1s and cDC2s in the spleen, suggesting enhanced differentiation of cDC progenitors. Finally, cDC survival was considered. cDCs from IL-2cx-treated versus untreated mice demonstrated no differences in apoptotic markers or regulators, despite a modest survival benefit after adoptive transfer. Taken together, IL-2-mediated cDC expansion likely occurs primarily through mature cDC proliferation and cDC progenitor differentiation.
Next, the authors considered how IL-2 therapy contributes to cDC expansion on a molecular level. This would occur most directly if cDCs could signal through the IL-2 receptor, in either a dimeric IL-2Rβγ or a trimeric, high-affinity form including IL-2Rα (CD25). Transcriptome analysis showed that cDCs expressed IL2rg but not IL2ra/b mRNA, in contrast to Tregs, which expressed all three. This result was confirmed through cell surface staining, in which DC progenitors did not express CD122 and CD25. Subsequently, the researchers aimed to directly assess IL-2R signaling through phosphorylation of STAT5, a downstream effect of IL-2/IL-2R (and CSF2) signaling. CSF2 (as a positive control), but not IL-2, induced pSTAT5 in cDCs and cDC precursors; in contrast, Tregs readily phosphorylated STAT5 upon IL-2 exposure. These results suggest that cDCs do not express functional IL-2 receptors. Human cDCs likewise expressed CD132, but not CD25 and CD122, and IL-2 stimulation similarly failed to generate pSTAT5. To further confirm that cDC expansion after IL-2 therapy is not due to direct engagement with IL-2Rs on cDCs, the researchers generated chimeric mice containing an equal mixture of wild-type and IL-2rg-/- DCs. Following IL-2cx treatment, both DC populations expanded equivalently, regardless of IL-2R expression.
Having shown that cDC expansion was not mediated directly through IL-2 signaling, the authors considered which intermediaries may be responsible. FLT3L and CSF2 are known to maintain DC homeostasis and proliferation. However, IL-2 still robustly expanded cDCs in Flt3l-/-, CSF2-/-, and Flt3l-/-CSF2-/- mice, implicating additional mediators. Previously, IL-2cx was observed to induce tumor necrosis factor (TNF), and the researchers hypothesized TNF signaling may be involved in cDC expansion. As predicted, blocking TNF using a soluble TNFR2-Fc-IgG1 fusion protein inhibited IL-2cx-mediated cDC expansion in Flt3l-/-CSF2-/- and wild-type mice. Additionally, IL-2cx more significantly expanded wild-type over TNFR-knockout cDCs in a bone marrow chimera, further implicating direct TNF signaling in DC expansion. Increases in serum TNF and FLT3L were observed in IL-2cx-treated mice and recombinant IL-2-treated patients (compared to pre-therapy). Overall, these observations highlight TNF, FLT3L, and CSF2 as likely intermediaries for DC expansion.
Next, Raeber et al. interrogated which cells directly engage with IL-2 and produce these factors. As chimeric mice reconstituted with wild-type, but not IL-2rg-/-, bone marrow could expand splenic cDCs after IL-2cx therapy, the cell source was determined to be hematopoietic rather than stromal. More specifically, IL-2cx boosted proliferation and the abundance of splenic CD4+ and CD8+ T cells, NK cells, and innate lymphoid cells (ILCs). Each of these lymphoid subsets expressed Flt3l, Csf2, and Tnf mRNA in the steady state. In some cases expression of these mRNAs increased with in vitro IL-2 stimulation, as did intracellular staining for CSF2 and TNF proteins. NK cells and ILCs appeared to be especially important, as mice lacking T and B cells, or reconstituted with type-2 ILC precursors, enabled IL-2cx-mediated cDC expansion, but ablating NK cells and/or ILCs diminished this effect. These results suggest lymphoid cells as a major driver of cDC expansion following IL-2 therapy.
Finally, the researchers investigated these phenomena in a therapeutic context. cDCs from IL-2cx-treated mice upregulated activation markers (CD40, CD80, CD86 and class I MHC) and expressed genes associated with the complement system, cytotoxicity, and CD103. Compared to cDCs from untreated mice, these cDCs were superior at OVA antigen uptake in vitro and OT1 stimulation in vivo. In the anti-PD-1-refractory B16F10 melanoma model, IL-2cx treatment increased intratumoral cDC1s and cDC2s and delayed tumor growth, which could be abrogated by depleting cDCs or CD11c+ cells. Characterizing TCGA human melanoma tumors, the researchers noted extended survival of patients with a high signature of IL-2 or BATF3, a cDC1 transcription factor. In fact, these signatures, along with BATF3, correlated with a lymphocyte signature, CSF2, FLT3L, TNF, and IL-2R subunits.
In summary, Raeber et al. uncovered a link between IL-2 immunotherapy and cDC responses, which may underlie the groundbreaking long-term remissions observed in IL-2-treated patients. IL-2 engages with a variety of lymphoid cells, specifically CD4+ T cells, CD8+ T cells, NK cells, and type 1-3 ILCs, which produce FLT3, CSF2, and TNF. These molecules interact with mature DCs and DC progenitors , promoting expansion, activation, and T cell priming. These results reveal how even decades-old immunotherapies can be supplemented by recent discoveries in immune cell biology, such as cDC biology and ontogeny, to uncover mechanistic insight and inform therapeutic strategies.
Write-up by Alex Najibi, image by Lauren Hitchings
[1] Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol 2014.
Meet the researcher
This week, first author Miro Raeber answered our questions.

What prompted you to tackle this research question?
Not only has the discovery of checkpoint inhibitors added a new pillar to cancer treatment, but the introduction of anti-PD-1 and anti-CTLA-4 antibodies has also fundamentally changed clinical oncology. Nevertheless, a large proportion of patients fail to respond to that new class of drugs. For an example, studies have suggested that so-called “cold” tumors with poor immune infiltration are less responsive to checkpoint inhibitors than highly infiltrated or “hot” tumors. Thus, a major question in current cancer research is how cold tumors can be transformed into hot ones. Working on interleukin-2 (IL-2) and improved IL-2 formulations (e.g., IL-2 complexes) in the laboratory of Onur Boyman while earning my master’s degree made me realize that although the first approved immunotherapy for cancer was introduced nearly 30 years ago, how it precisely orchestrates antitumor immune response remains unclear. Especially because IL-2 acts via a different axis than checkpoint inhibitors, it may be an outstanding candidate for future combination treatments, and, for that reason, investigating IL-2’s novel aspects has been fascinating.
What was the most surprising finding of this study for you?
Perhaps most surprising was an increase in dendritic cells (DCs) after IL-2 treatment, for we knew that DCs do not express functional IL-2 receptors. While elaborating the detailed mechanism of IL-2-mediated DC expansion, we observed that not only T and natural killer cells, but also innate lymphoid cells (ILCs), contributed to the production of DC-stimulating cytokines. However, without an ILC depletion model available, we had to develop different strategies for conclusively showing the contribution of ILCs to IL-2-mediated DC expansion. After numerous failed attempts, we finally managed to reconstitute Il2rg-deficient mice with IL-2-responsive ILCs via the adoptive transfer of highly purified ILC precursors, which showed their ability to expand DCs independently. Beyond that, it was remarkable to observe how IL-2 complexes expand intratumoral DCs so profoundly, whereas anti-PD-1 treatment barely affects them.
What was the coolest thing you’ve learned (about) recently outside of work?
While reading Sapiens by Yuval Noah Harari, I first learned about gossip theory, which generally holds that humans are social animals whose success as a species depends heavily upon social cooperation. To efficiently track social relations and, for example, who can be trusted or not, humans have learned to gossip effectively and thus distinguished themselves from other apes. In that sense, we are all merely gossiping apes, which is an entertaining thought.



