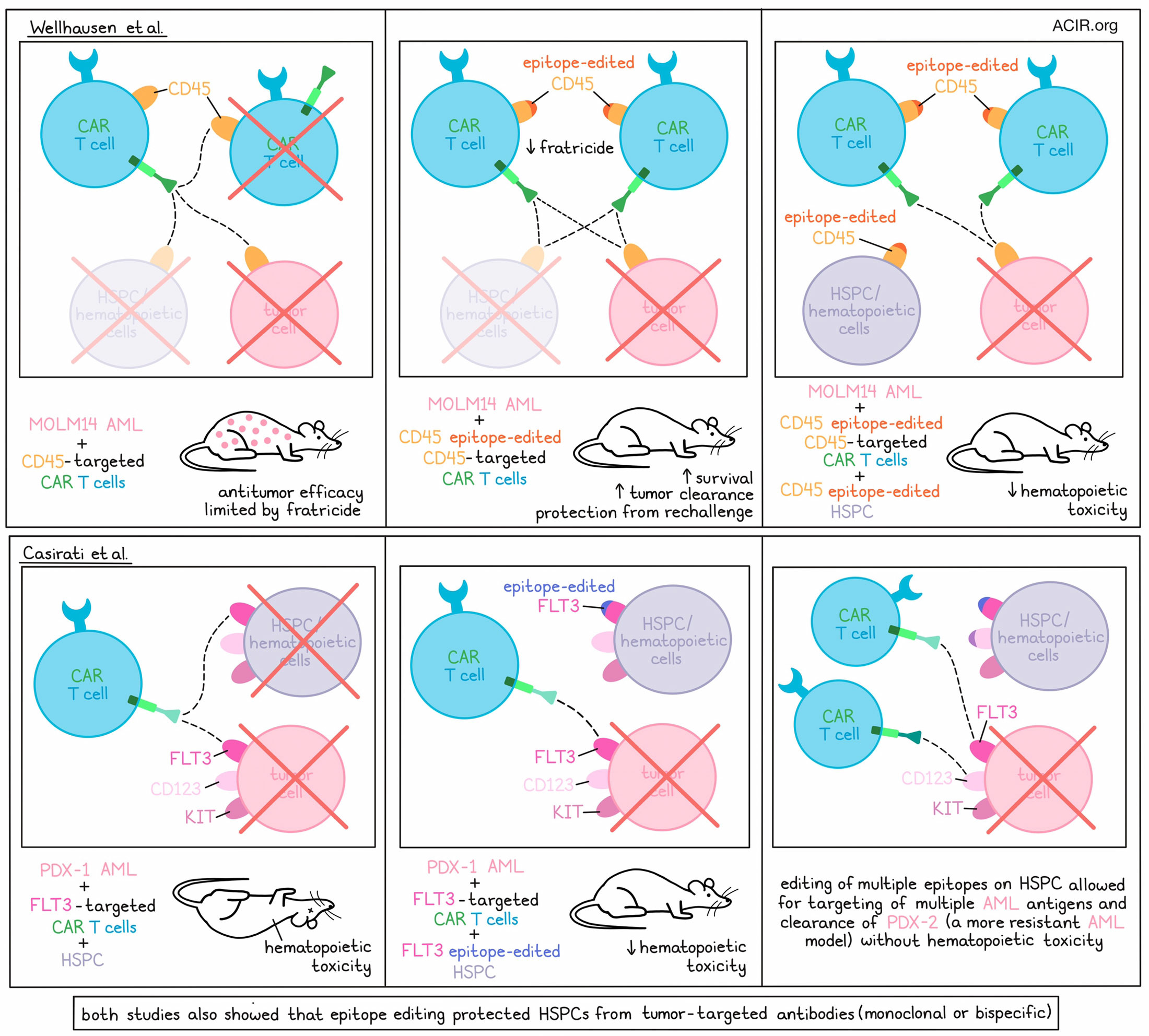
CAR T cell therapy for hematological cancers can be highly effective, but is currently limited to antigens that are only expressed by tumor cells or by lineages that are non-essential. Targeting antigens expressed by all tumor cells is often associated with on-target/off-tumor toxicities to normal hematopoietic cells. Two recent papers used epitope editing to overcome this issue. The work by Wellhausen et al. on the pan-hematologic CD45 epitope editing was published in Science Translational Medicine and the research by Casirati et al. on editing of three acute myeloid leukemia (AML) antigens was published in Nature.
Wellhausen et al. aimed to target CD45, as it is expressed by most hematological cancer cells. However, CD45 is expressed on all hematopoietic stem cells and their progeny, including the CAR T cells themselves, and is required for their function. Therefore, even though CD45 CAR T cells (CAR45) would target leukemias and lymphomas, they would be limited by fratricide, and would result in pancytopenia due to on-target/off-tumor toxicity. CAR45 constructs were created by cloning anti-CD45 single-chain variable fragments (scFvs) from a selected antibody clone, BC8, which recognizes CD45, into second-generation CAR lentiviral vectors. However, as expected, these CAR45 led to a reduction of T cell numbers due to fratricide.
To overcome fratricide, the researchers aimed to identify a non-synonymous mutation at a CD45-relevant epitope using CRISPR base editing, so that CAR45 would not recognize it, while simultaneously maintaining the expression and function of CD45. Initially the researchers expressed myc-tagged CD45RO constructs that sequentially truncated the ectodomain from the N-terminus in CD45- NALM6 cells. This revealed that the binding site for BC8 was the D1 domain.
To map the amino acids required for the binding of CAR45 constructs, the researchers performed alanine mutagenesis of the CD45 D1 domain, again using myc-tagged WT CD45RO as a template. Candidate targets were selected based on differences between mouse and human. Alanine was substituted for each candidate amino acid in CD45, and mutant CD45 constructs were expressed in NALM6 cells. None of the mutations affected CD45 expression. When NALM6 cells with CD45 alanine mutants were cocultured with CD45KO CAR primary human T cells, four mutation constructs reduced or removed BC8-derived CAR T cell activation. Superimposing the amino acids relevant for binding to the BC8 CAR onto the crystal structure of the CD45 extracellular domain revealed a conformational epitope.
To introduce the mutation in the epitope on CD45 targeted by CAR45, CRISPR base editing was used to install base pair changes at the targeted loci. Two gRNAs were identified that resulted in a single amino acid substitution at the CD45 epitope, which resulted in a complete lack of CD45 binding by the BC8 antibody. Transduction of CAR45 into a mixed population of T cells with or without epitope editing of CD45, resulted in the elimination of non-edited cells and enrichment of edited cells. The CAR45 cells were able to kill CD45WT-expressing cells, while they could not kill CD45BE (base-edited) cells.
To determine the antitumor efficacy of CAR45BE, acute myeloid leukemia (AML) was used as a model. The CAR45BE could kill MOLM14 AML cell lines in vitro, and in vivo experiments with primary AML samples in NSG-SGM3 mice resulted in the eradication of AML within 3 weeks after injection of the CAR45BE, resulting in long-term survival. The CAR45 cells proliferated in vivo and contracted after clearance of the tumor cells. Mice remained tumor-free after rechallenge with the same primary AML cells after 3 months.
However, this CAR-T construct would lead to on-target/off-tumor toxicities against healthy hematopoietic cells, which express CD45. To overcome this, the researchers used the same epitope base editing in human CD34+ hematopoietic stem and precursor cells (HSPC), to protect them from CAR45BE killing. The HSPC could proliferate and differentiate in vitro, and engraftment into NSG mice resulted in immune reconstitution. Edited HSPC were able to differentiate into major immune cell subtypes similar to unedited HSPC, and the immune functions of these subsets were maintained.
To determine whether these epitope-edited HSPC and their progeny were resistant to killing by CAR45BE, NSG mice were engrafted with epitope-edited CD34+ HSPC, injected with MOLM14 AML cells, and treated with CAR45BE. Therapy resulted in a decrease in tumor burden and improved survival, while human HSPC remained in the peripheral blood and bone marrow, suggesting they were protected from killing.
Finally, the researchers hypothesized that epitope editing could also protect cells from bispecific T cell engagers (BTE) targeting CD45. In a similar NSG model with MOLM14 AML tumor cells and epitope-edited T cells, injection with a BTE targeting CD3 and CD45 resulted in tumor clearance and survival improvement. In vitro experiments confirmed that epitope-edited T cells with CD45BE-expressing NALM6 cells were not lysed after BTE treatment, suggesting the epitope editing strategy could also be combined with BTE treatment.
Casirati et al. took a more HSPC-edited target-specific approach and generated base-edited versions of three potential AML antibody targets – FLT3, KIT, and CD123 – to generate “stealth receptors”, which lost antibody binding while preserving physiological functions. Similar to the results with CD45 base editing, epitope engineering of these receptors was feasible and resulted in normal functioning in vitro, and K562 cells engineered to express the stealth receptor were resistant to killing by second-generation CAR T cells carrying scFvs targeting the unedited epitopes.
Human HPSC were therefore base-edited for the binding site of any one of the three clones. The edited cells had no skewing of the composition of phenotypically identified progenitor cells, and showed a similar functionality to non-edited cells. The HSPCs did not have differences in receptor signaling or stem cell differentiation ability, and were similar to controls in terms of the functionality of their lineage-specific progenies. Stealth receptor-modified HPSCs were resistant to killing by CAR T cells carrying cognate scFvs (for FLT3 of CD123) or to killing by monoclonal antibody (for KIT; antibody therapy was assessed for KIT because of the known extra-hematopoietic expression of KIT and the hypothesis that antibody therapy would be less toxic than CAR therapy).
The researchers then assessed whether targeting the unedited epitopes could kill AML cells while maintaining base-edited HPSC. In vitro killing assays showed that cells with unmodified FLT3 or CD123 were killed by CAR T cells targeting the unedited epitopes, while cells with the epitope-engineered variants were resistant to killing.
To determine whether in vivo CAR T cell therapy could eliminate AML cells while preserving edited HSPCs, the researchers used a human patient-derived AML xenograft (PDX-1), which has a FLT3 mutation, in mice engrafted with the FLT3 epitope-edited HPSCs. Mice were treated with FLT3-targeting CAR T cells, resulting in AML cells eradication in the bone marrow and spleen, while HSPCs were protected. These experiments were repeated with the CD123-targetingg CAR T and CD123 epitope-edited cells, which also showed antitumor efficacy and a reduction of hematopoietic toxicity.
Given the heterogeneous nature of AML, the researchers hypothesized that editing and targeting two or more epitopes in HSPCs might enable more effective immunotherapy. To assess this, they co-edited two (FLT3 and CD123) or three (FLT3, KIT, and CD123) targets on K562 cells and cocultured these cells with bi- or tri-specific CAR T cells. Only the edited cells survived CAR T cell-mediated killing and did not induce T cell activation, degranulation, proliferation, or cytokine secretion. In the PDX-2 AML model, which was partially resistant to eradication by a CAR T targeting FLT3, treatment with this FLT3-targeting CAR T plus a CAR T targeting KIT or CD123 was more effective. To confirm that the dual-edited HSPC were resistant to this therapy, dual epitope-edited (FLT3 and CD123) were engrafted into NBSGW mice, after which mice were injected with the PDX-2 cells and treated with FLT3- and CD123-targeting CAR-T cells. This therapy eradicated the AML cells from the bone marrow and spleen, while the epitope-edited cells persisted. These data suggest that multiplex epitope editing can be performed in HSPCs to overcome the toxicities of multitarget immunotherapies.
The exciting data from these two papers opens the door to new strategies to improve the efficacy of targeted therapies in patients and expand the number of targetable patients. If the engraftment of epitope-edited HSPC can be performed safely in humans, potentially more effective immunotherapies targeting antigens ubiquitously present on tumor cells and healthy hematopoietic cells could be assessed.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Gabriele Casirati and lead author Pietro Genovese answered our questions.

What was the most surprising finding of this study for you?
When we set out to identify engineered variants of surface molecules, we did not anticipate that single amino acid changes (actually, single base changes!) would be enough to disrupt antibody and CAR binding. Even more surprising, we could reproduce faithfully these mutations through adenine base editing.
What is the outlook?
We are now working to further optimize our editing protocol to achieve an even higher degree of precision for clinical translation. We plan to prepare the data for an Investigational New Drug (IND) in the next 12-18 months, with the goal of initiating an investigator-sponsored first-in-human trial for patients with relapsed/refractory Acute Myeloid Leukemia.



