Treatment of solid tumors with CAR T has various barriers to overcome to be effective. To improve efficacy, boosting the immune response by delivering pattern recognition receptor (PRR) agonists more specifically to immune than tumor cells might improve responses, given the known role of some PRRs to drive tumor progression. To do this, Johnson et al. engineered a CAR that delivers the RNA RN7SL1 preferentially to immune cells, and tested its use and effects on endogenous immune responses in multiple animal models. Their results were recently published in Cell.
Johnson et al. designed two RNAs, RN7SL1 and a control consisting of a scrambled sequence of RN7SL1 (Scr). These RNAs upregulate interferon (IFN)-stimulated genes, including MHC-I, PD-L1, and CD86 on B16F10 melanoma cells and human DCs. This effect is dependent on RIG-I and MDA5 for RN7SL1, while only one of those PRRs is required for Scr. To test its effects in vivo, mice with B16 tumors were intratumorally injected with liposome-encapsulated RNA. When mice were depleted of T cells using antibodies, RN7SL1, not Scr, enhanced tumor growth and decreased survival in mice lacking T cells. In contrast, when mice were treated with checkpoint inhibitors (anti-PD-1 + anti-CTLA-4) in the presence of T cells, RN7SL1, but not Scr, improved checkpoint blockade responses, with increased T cell activation and DC infiltration.
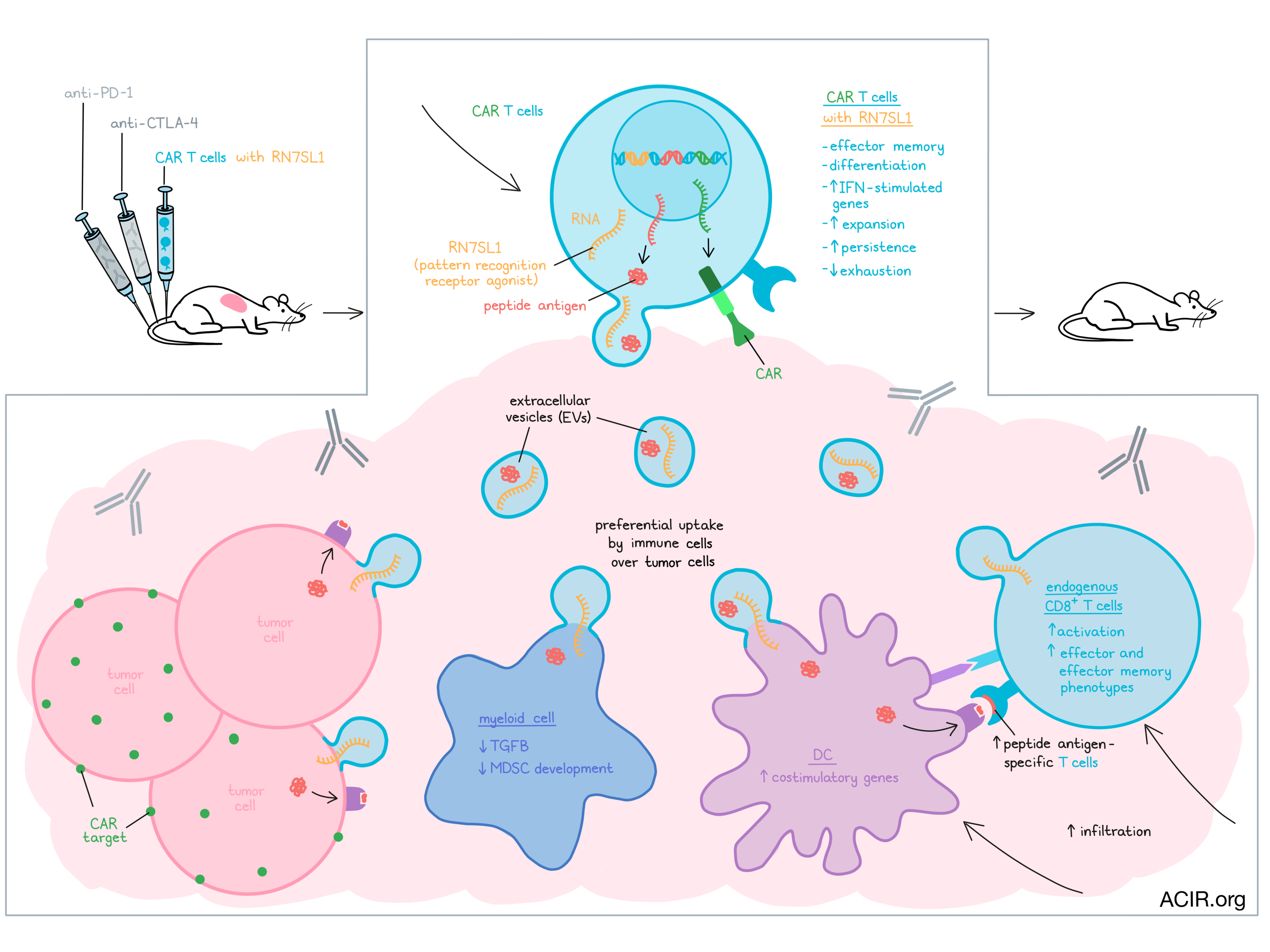
To ensure that RN7SL1 would preferentially be activated in immune cells, and less in cancer cells, the researchers hypothesized that the IFN activating effects of CAR T cells in a tumor might lead to the preferential delivery of RN7SL1 co-expressed in those CAR cells. Human and mouse CAR T cell models were used to assess this approach with mesothelin (M5BBz)- and CD19 (19BBz)-specific CAR T cells. An RNA polymerase III-dependent U6 promoter downstream of the CAR sequences was used to drive the transcription of RN7SL1 or Scr. The researchers first assessed the effects of expressing RN7SL1 on the CAR T cell itself. After CAR T expansion, those co-expressing RN7SL1 were more likely to be effector-memory cells. Treatment of mice bearing human mesothelin-expressing ASPC-1 pancreatic tumors with the M5BBz CAR T showed that the CAR T co-expressing the RNA (M5BBz-7SL) had an increased efficacy, resulting in durable responses. When tumors were assessed by single-cell RNAseq, the infiltrated M5BBz-7SL CAR T were also predominantly of a memory T cell type and showed higher expression of type I IFN-stimulated genes than the M5BBz CAR T cells, which appeared to have an exhausted phenotype. Additionally, the M5BBz-7SL CAR T expanded and persisted in the tumor and blood, with peripheral CAR T being mainly of the effector-memory phenotype.
Based on their previous work showing that RN7SL1 can be transferred between cells through exosomes, Johnson et al. hypothesized RN7SL1 might be exported in extracellular vesicles (EVs) to preferentially activate IFN signaling in immune cells, rather than tumor cells. Confirming this hypothesis, RN7SL1 and Scr were found in purified EVs secreted by CAR T cells as well as in endogenous immune cells after treatment, including DCs and T cells, an effect that was not seen when EV secretion was inhibited.
Given this transfer to immune cells, the researchers hypothesized that the antitumor effects of RN7SL1 might be more extensive than only its autonomous effects on the CAR T cells. Mice were treated with the CAR T combined with anti-CTLA-4 or anti-PD-1 and this resulted in increased tumoral CAR T and endogenous immune cell infiltration, and improved outcomes. Both RN7SL1 and Scr RNA increased expression of IFN-stimulated genes in CAR T and endogenous immune cells. Scr RNA delivery resulted in an accumulation of myeloid cells expressing MDSC genes, while RN7SL1 delivery resulted in decreased levels of Tgfb1 and limited MDSC accumulation. For DCs, RN7SL1 resulted in increases in CD209a+ pDC-like cells and promoted changes in regulatory gene modules expressed by the DC1 subset, resulting in enrichment of co-stimulation genes. These data suggest positive effects on the balance between anti- and pro-tumoral responses of myeloid and DC subsets in the tumor microenvironment. Importantly, a higher infiltration of endogenous CD8+ T cells was observed in treated tumors, and RN7SL1 delivery increased the frequency of effector-like and effector-memory-like T cells.
When Tcra or Batf3 knockout mice (lacking T cells or DC1s, respectively) were implanted with B16-h19 tumors and treated with CAR T expressing RN7SL1, the effects on survival were limited. These data suggest that the impact of RN7SL1 relies on CD8+ T cells that are primed by DCs and recruited to the tumor. Additionally, these immune effects were reliant on IFN-I signaling, since in Rig-1-/- mice and mice treated with anti-IFNAR antibodies, the beneficial effects were limited.
The researchers observed an increase in TRP2-specific CD8+ T cells (an immunogenic antigen in B16F10 tumors) expressing higher levels of GZMB and Ki67 in tumors treated with the CAR T expressing RN7SL1. Therefore, loss of the CAR T target might not result in resistance as it induces endogenous antigen-specific T cell responses. To investigate this, mice implanted with a 1:1 mix of CD19+ and CD19- B16 tumor cells were treated. Mixed tumors were eradicated similarly to the homogenous CD19-expressing tumors, and upon rechallenge with CD19- B16 tumors, most mice remained tumor-free, suggesting induction of memory responses.
An additional issue restricting the efficacy of CAR T therapy for solid tumors is limited neoantigen expression in many solid tumors. To address this, the researchers engineered CAR T cells that deliver not only RN7SL1 but also antigens, with the idea to ‘paint’ the tumor cells with exogenous antigens. CAR T expressing the SIINFEKL peptide alone or with RN7SL1 were cultured with B16 tumor cells, which induced a dose-dependent increase in SIINFEKL presentation by MHC-I on the cancer cells that could activate OT-I T cells (specific for SIINFEKL). In vivo, Ki67+ Ova-specific CD8+ T cells infiltrated treated tumors, increasing the treatment’s efficacy. In a low mutational burden tumor (KP lung tumors heterogeneously expressing CD19), transfer of OT-I T cells (to deliver a T cell pool) and treatment with CAR T delivering both SIINFEKL and RN7SL1 significantly delayed tumor growth, an effect not seen with antigen or RN7SL1 alone.
These data suggest that arming CAR T with RN7SL1 may improve endogenous T cell responses towards tumors, increase antitumor efficacy, and prevent therapy resistance. Additionally, the co-delivery of antigens may further enhance the efficacy of treatment in tumors with limited immunogenic antigen expression. Thus, such strategies may overcome some of the challenges regarding the use of CAR T cells to treat solid tumors.
Write-up by Maartje Wouters, image by Lauren Hitchings



