Immune checkpoint blockade (ICB) targeting PD-1 is increasingly used as neoadjuvant therapy. However, the mechanisms and cell types determining ICB responses remain poorly understood. Oliveira, Egloff, Afeyan, et al. compared T cell activation status and dynamics in responders and non-responders to neoadjuvant pembrolizumab in patients with head and neck squamous cell carcinoma (HNSCC) and recently published their data in Science Immunology.
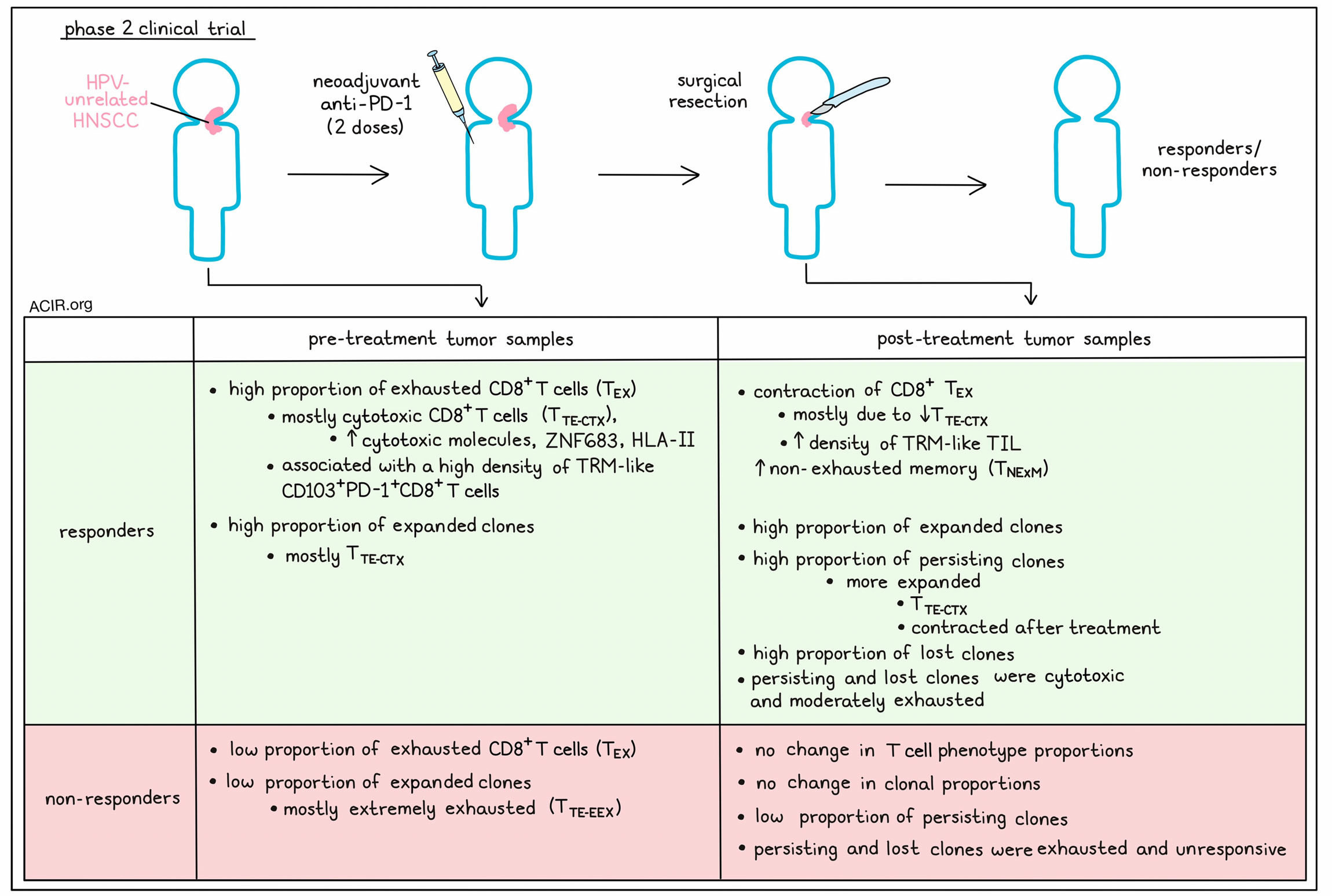
Samples for this study were obtained from 30 patients participating in a Phase 2 trial with HPV-unrelated HNSCC, who received two doses of pembrolizumab before surgery. Pathologic tumor responses were detected in 15 patients (52%).
To determine how T cell dynamics in the tumor microenvironment (TME) related to response to pembrolizumab, the researchers performed single cell RNAseq and TCRseq on CD3+ TIL from 14 tumor biopsies collected before and/or after pembrolizumab treatment. Samples from seven patients who had received two doses and one patient who received one dose were analyzed; four of the patients were responders. Pre- and post-anti-PD-1 biopsies were available from 6 patients (3 responders and 3 non-responders).
There were 12 clusters of CD8+ TIL, and the most abundant terminally exhausted (TTE) cell subpopulations could be subdivided into two subclusters. One subcluster was characterized as extremely exhausted (TTE-EEX), with differentially expressed genes related to exhaustion and recognition of tumor antigens. The second subcluster had cytotoxic characteristics (TTE-CTX) and appeared less severely exhausted. Both subclusters had high expression of tissue residency genes, but differed in terms of transcription factors. TTE-CTX had the highest expression of cytotoxic molecules (perforin, interferon and granzymes) and the ZNF683 transcription factor – which has been associated with the maintenance of the tissue-resident memory (TRM) program). TTE-CTX also upregulated HLA-II genes, consistent with effector-like activation.
To investigate how these subsets related to antigen recognition, the researchers projected TCR clonality to the phenotypes of CD8+ TIL. CD8+ TCR clonotype families mostly clustered into non-exhausted memory (TNExM) or exhausted (TEX) clusters. TNExM clusters were enriched for signatures related to viral antigen specificity, while TEX had profiles previously associated with tumor-reactive TIL. These putative tumor-reactive TIL resembled terminally differentiated TIL and included a small percentage of TIL with enriched progenitor-exhausted signatures.
To relate the T cell dynamics with therapy response, the researchers compared responders with non-responders. Responders had higher frequencies of CD8+ TEX TIL in pre-treatment samples, of which most were TTE-CTX. Comparing matched pre- and post-treatment biopsies from six patients revealed that five weeks after treatment started, the three responders had a contraction of the CD8+ TEX compartment, with an increase in the TNExM population, while non-responders did not show apparent differences. Most of the contraction in responders was due to a reduction in CD8+ TTE-CTX.
Responders had a higher proportion of CD8+ TEX TIL with cytotoxic potential before treatment, which was associated with a higher density of TRM-like CD103+PD-1+CD8+ TIL in pretreatment samples by immunofluorescence, an effect that was stronger in better responders. The density of these TRM-like TIL increased slightly after therapy, but remained higher in responders than in non-responders.
The researchers validated the data using independent cohorts. Analysis of bulk RNAseq of tissue obtained from 26 patients participating in a previous one-dose pembrolizumab trial showed more CD8+ infiltration in responders, and enrichment in baseline expression of ZNF683 and CTX genes (related to TTE-CTX). An external dataset with bulk tumor RNAseq data of 11 patients treated with three doses of nivolumab also confirmed that ZNF683 and the CTX signature were enriched in responders pre-treatment. These data suggest responses to ICB depend on high levels of pre-existing exhausted/cytotoxic TIL.
The researchers then determined the dynamics of individual TCR clonotypes pre- and post-treatment. In responders, the majority of pre-treatment CD8+ TEX TILs were highly expanded clonotypes with a TTE-CTX phenotype, while non-responders had fewer expanded CD8+ TEX TCR clonotypes, which were mostly TTE-EEX. To examine whether pre-existing TCR clonotypes persisted or there was an influx of new T cells, TCR clonotypes were categorized as only present in pretreatment samples (lost), only present in post-treatment samples (new), or present in pre- and post-treatment samples (persisting). There were numerous new TCR clonotypes after treatment, but most were not expanded and likely contributed little to the antitumor repertoire post-treatment in both responders and non-responders. Many clonotypes detected at baseline persisted after treatment (60-70% of all CD8+ TIL), responders had larger proportions of persisting clones, and persisting clones were more expanded post-treatment. In responders, the persisting and expanded CD8+ TEX clones contracted after treatment; therefore, responders had a high proportion of clones that were lost after treatment, while non-responders did not have changes in TEX clonal proportions.
In the CD4+ T cell population, TNExM, TEX, GZMK+ T and Tregs were detected. TEX and TGZMK were enriched for signatures of validated tumor-specific TCRs. In responders, there was a similar enrichment in the CD4+ population of exhausted cells, as in the CD8+ population, while non-responders had higher frequencies of CD4+ TNExM. Proportions of Tregs were similar in responders and non-responders.
Among all lost, new, and persisting TNExM and TEX TCR clonotypes, the putative tumor-reactive TEX TCR clonotypes showed the most therapy-induced differences. The highest levels of response-associated transcripts, such as for cytotoxicity genes, were expressed by cells that had persisting or lost TCR clonotypes in responders, and these TIL also had the highest cytotoxicity scores and moderate levels of exhaustion. On the other hand, persisting TEX clones in non-responders were highly enriched for transcripts associated with a lack of response (exhaustion genes), and expanded clones in these patients had the highest levels of exhaustion. The level of exhaustion among putative tumor-reactive CD8+ TEX clonotypes did not decrease in either responders or non-responders, and these cells did not acquire a memory phenotype. Tracking the responder-associated clonotypes showed these cells constituted about 55% of CD8+ TEX and about 32% of all CD8+ TIL in pre-treatment samples of responders. Highly expanded clones expressed ZNF683 and CTX genes, while in non-responders, this population only comprised about 4% of all CD8+ TIL. The population expressing ZNF683 and CTX genes declined most post-treatment in responders, through contraction of these clones. Non-responders, on the other hand, mostly had pre-existing TEX TCR clonotypes that were in a state of extreme exhaustion, which did not change after treatment.
These data show that responses to anti-PD-1 therapy in HNSCC are likely induced by CD8+ TEX TIL characterized by a strong cytotoxic and resident-memory signature that are present in the HNSCC TME pre-treatment. This novel observation differs from mechanistic studies in other diseases, which identified intratumoral expansion of less exhausted cells (clonal revival) and infiltration of novel clonotypes (clonal replacement). The results of this study could aid in determining which patients might benefit most from ICB treatment and provide clues as to why subsets of patients do not respond.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-first author Alex Afeyan answered our questions.

What was the most surprising finding of this study for you?
One of the most surprising findings was that in the patients we analyzed, response to immunotherapy seemed to be predetermined by the status of tumor infiltrating T cells. Indeed, the major driving factor of response to PD-1 blockade within the timeframe of our study seemed to be the revival of cytotoxicity of tumor-infiltrating CD8+ T cell clones present prior to therapy. Previous studies have highlighted two mechanisms for anti-PD-1 response, either the recruitment of new antitumor T cell clones to the tumor (clonal replacement), or the revival of exhausted T cell clones towards a less exhausted progenitor state (clonal revival). Instead, in head and neck cancers, we saw that responders were enriched in pre-existing expanded T cell clones that maintained a tissue-resident, cytotoxic phenotype, but underwent “cytotoxic revival”, unleashing their antitumor potential in responders to therapy.
What is the outlook?
Through this study comparing responders and non-responders, we identified a population of tumor-infiltrating CD8+ T cells expressing markers of cytotoxic potential and tissue residence (ZNF683+ CTX+ TILs) that significantly predicted and appeared to drive response in head and neck squamous cell carcinoma. We believe that our single-cell transcriptomic signatures of this population could be used in the future to guide therapy through stratification of patients by likelihood of response to anti-PD-1 therapy. Additionally, our analysis of external datasets suggested that the mechanism of cytotoxic revival may be active in other tumor types as well. Additional studies are needed to identify what tumor features drive the mechanisms of response and why the tumor microenvironment of non-responder patients is not able to support the expansion of ZNF683+ CTX+ TILs in favor of severely exhausted and dysfunctional T cells.
What was the coolest thing you’ve learned (about) recently outside of work?
As a lifelong Boston sports fan, I have learned a lot in the past several years about perseverance through (very low stakes) heartbreak, and our ability to bounce back and support our teams. I spent most of my life watching team after team bring home Superbowls, World Series, NBA Finals, and Stanley Cups. To the joy of many outside New England, the past few years have seen a particularly high number of losses late in the postseason, but just as many subsequent seasons where our teams buckle down and stick together, fighting the good fight and giving hope to many fans around the region. Go Celtics, Pats, Bruins and Sox!



