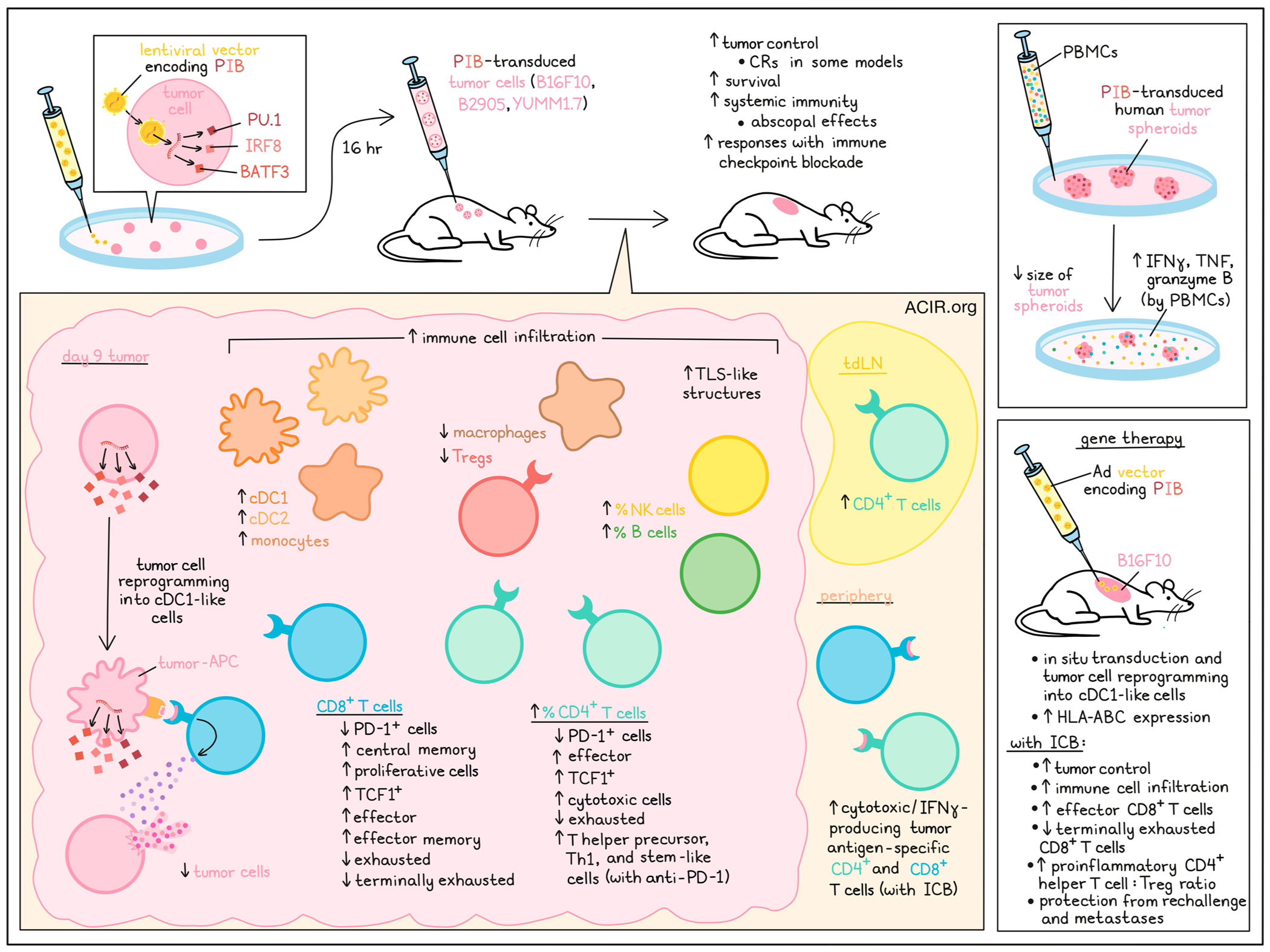
Type 1 conventional dendritic cells (cDC1s) are required for T cell-mediated tumor regression and response to immune checkpoint blockade (ICB) in cancer. However, current immunotherapy strategies are not focused on exploiting key aspects of cDC1 functionality in vivo, such as chemokine secretion and antigen cross-presentation. In a recent Science publication, Ascic et al. explored in vivo cellular reprogramming of tumor cells to obtain and maximize cDC1 functionality.
Previously, the researchers identified a combination of transcription factors (PU.1, IRF8, and BATF3 – named PIB) delivered by a lentivirus that can reprogram fibroblasts or tumor cells into cDC1-like cells in vitro. These reprogrammed cells expressed costimulatory molecules, secreted chemokines and cytokines, and activated T cells.
To explore whether PIB could also be used to drive in vivo reprogramming of tumor cells, the researchers made use of the poorly immunogenic murine melanoma cell line B16F10, which has low MHC expression and is resistant to ICB, and the immunogenic line B2905, which is a highly mutated melanoma tumor. To test the antitumor effects of in vivo cDC1 reprogramming, a mixture of 88% PIB-transduced B16F10 cells (16 hrs after transduction) and 12% non-transduced parental cells were implanted subcutaneously to establish tumors in mice. Thirty percent of the mice had complete responses (CR), and tumor growth was delayed in the other mice, extending survival. When this strategy was combined with double ICB (anti-PD-1 + anti-CTLA-4), all animals had tumor regression. The researchers confirmed that transcription factors were delivered to tumor cells and there was no phenotypic reprogramming before implantation in vivo. Bilateral tumor challenge experiments in which a mixture of transduced and parental tumor cells were implanted on one side, while only parental tumor cells were implanted on the other, showed that in vivo cDC1 reprogramming combined with ICB resulted in systemic immunity, as tumors on both sides regressed, and the number of cytotoxic melanoma antigen-specific T cells in the periphery increased.
The researchers then assessed in vivo cDC1 reprogramming as a monotherapy and in combination with PD-1 or CTLA-4-targeting therapy in the B16F10, B2905, and YUMM1.7 models (which is resistant to ICB and depends on cDC1 availability). Mice were implanted with a 1:1 mixture of transduced and parental tumor cells. Monotherapy resulted in 100% CRs in YUMM1.7, 80% in B2905, and increased survival in B16F10-challenged mice. The reprogramming synergized with anti-PD-1 or anti-CTLA-4, leading to increased CRs in the B2905 and B16F10 models, and the expansion of tumor-specific CD8+ and CD4+ IFNγ-producing peripheral T cells.
Examining the effects of the reprogramming on the tumor microenvironment (TME), the researchers analyzed tumors on day 9 and found that there was an increase in CD45+ cells and a decrease in tumor cells. Dense lymphocyte clusters resembling tertiary lymphoid structures (TLS) were observed in the tumors. The percentage of B cells, NK cells, and CD4+ T cells increased, and while there was no change in the percentage of CD8+ T cells, there was a decrease in PD-1+ CD8+ and CD4+ T cells. Furthermore, central memory CD8+ T cells, proliferating CD8+ T cells, effector CD4+ T cells, and TCF-1+ CD8+ and CD4+ T cells increased. Furthermore, the number of cDC1s, cDC2s, and monocytes increased, while the number of Tregs and macrophages decreased. In the tumor-draining lymph nodes (tDLNs), CD4+ T cells expanded, but no clear changes in other immune populations were observed.
Ascic et al. then profiled T cells by scRNAseq with T cell receptor (TCR) enrichment. For CD8+ T cells, increases in intratumoral effector and effector memory cells were observed in PIB-treated tumors, while exhausted and terminally exhausted populations decreased. There was a large cluster of cytotoxic CD4+ T cells in reprogrammed tumors, and the number of exhausted and regulatory CD4+ cells decreased. When combined with anti-PD-1, CD4+ T cells shifted toward T helper precursors, Th1, and stem-like fates.
To determine whether there was polyclonal expansion, the full-length αβTCR sequences with more than one cell per clonotype were assessed. Similar numbers of clonotypes of expanded CD8+ T cells were observed in reprogrammed and control tumors; however, in reprogrammed tumors, expanded clones were enriched for effector and effector memory subsets.
Next, the researchers determined whether human cancer cells could be reprogrammed into an immunogenic cDC1 phenotype in vivo. Human melanoma (A375 and A2058) and glioblastoma (T97G) tumors were transduced in vitro with the PIB vector and immediately implanted into NSG mice, where in vivo reprogramming occurred. At day 9, reprogrammed cells were detected in all three models, and the cells expressed the cDC1-specific surface markers XCR1, CLEC9A, and CD226, as well as increased HLA-I expression.
To assess the effects on the human TME, human cancer cell lines were transduced and spheroids were generated. scRNAseq of reprogrammed cells showed a fast upregulation of endogenous SP11, IRF8, and BATF3, reprogramming markers, and cDC1 genes. The presence of cancer-associated fibroblasts, myeloid-derived suppressor cells, or pericytes in the spheroids did not impact cDC1 reprogramming efficiency. When PBMCs were added to the culture, the reprogrammed spheroids reduced in size (while controls did not), and there was production of IFNγ, TNFα, and granzyme B by the PBMCs, supporting antigen presentation and T cell-stimulating capabilities in the transduced spheroids.
To establish what platform could be used to deliver PIB to tumors to elicit direct in situ cDC1 reprogramming, Ascic et al. compared lentiviral vector (LV), non-integrative and replication-deficient adenoviral (Ad), and adeno-associated viral vectors (AAV). The reprogramming efficiency and induced MHC-I expression were higher with Ad and LV than with AAV. Intratumoral injections showed high in situ transduction capacity for Ad and AAV, while the capacity of LV was low. In human xenograft models, Ad resulted in transduction, in situ reprogramming, and increased HLA-ABC expression.
To test this gene therapy approach, B16F10 tumors were established, and mice were injected intratumorally with Ad-PIB on days 7, 9, 11, and 13, combined with anti-PD-1 and anti-CTLA-4. Half of the mice had a CR and remained tumor-free long-term. There was immune cell influx into the tumors, with an increase in the proportion of effector and a decrease in terminally exhausted CD8+ T cells. The ratio of pro-inflammatory CD4+ T helper cells to Tregs also increased. Rechallenge experiments showed mice remained tumor-free. To assess the effects on metastasis, mice were injected IV with B16F10 cells, and survivor mice did not develop metastases.
In conclusion, this research confirms the ability of PIB to reprogram tumor cells into cDC1s in vivo, allowing for immune-cold tumors to become hot. If this method can be translated to the clinic, it might allow for improvements in immunotherapy responses.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, co-authors Ervin Ascic and Fritiof Åkerström answered our questions.

What was the most surprising finding of this study for you?
The most surprising finding for me was that generating an immunogenic cDC1 fate in cancer cells was not hindered by the immunosuppressive environment. In fact, we found evidence in some cancer types that the reprogramming process was actually enhanced by the presence of immunosuppressive cells, leading to more efficient generation of cDC1-like cells. Current cancer immunotherapies, such as checkpoint inhibitors or CAR T cells, often struggle due to the immunosuppressive tumor microenvironment. However, this was not the case for in vivo cDC1 reprogramming, which was particularly exciting!
What is the outlook?
At the preclinical level, we will further optimize the viral vectors to ensure patients receive the most advanced gene therapy. We will also conduct additional safety studies, including rigorous assessments of dosing and toxicity. The overarching goal is to reach first-in-human trials, where we aim to test the concept in patients with untreatable solid cancers. Initially, we will assess the safety and efficacy of this approach as a monotherapy, as well as in combination with immune checkpoint inhibitors. As this study has demonstrated the potential for recreating immune cells in vivo, another future step is to explore generating other immune cell subsets that could be used to treat different indications.
What was the coolest thing you’ve learned (about) recently outside of work?
Ervin Ascic: Outside of the lab, I’m passionate about cooking. I love experimenting with new ideas that I pick up from my travels around the world. Cooking, like lab work, requires following protocols, understanding where you can experiment, and embracing trial and error. For example, when I was perfecting my homemade pizza dough recipe, it kept challenging me until I finally optimized it and could proudly share it with friends and family. Patience and creativity are key, and when everything clicks, the result is incredibly rewarding.
Fritiof Åkerström: One of my favorite hobbies is enjoying outdoor activities, particularly fishing and hiking. Recently, I went hiking in Scotland, and it truly lived up to its reputation as one of the most scenic destinations in the world. These outdoor activities allow me to disconnect from the lab, recharge, and gain fresh perspectives to tackle new challenges.



