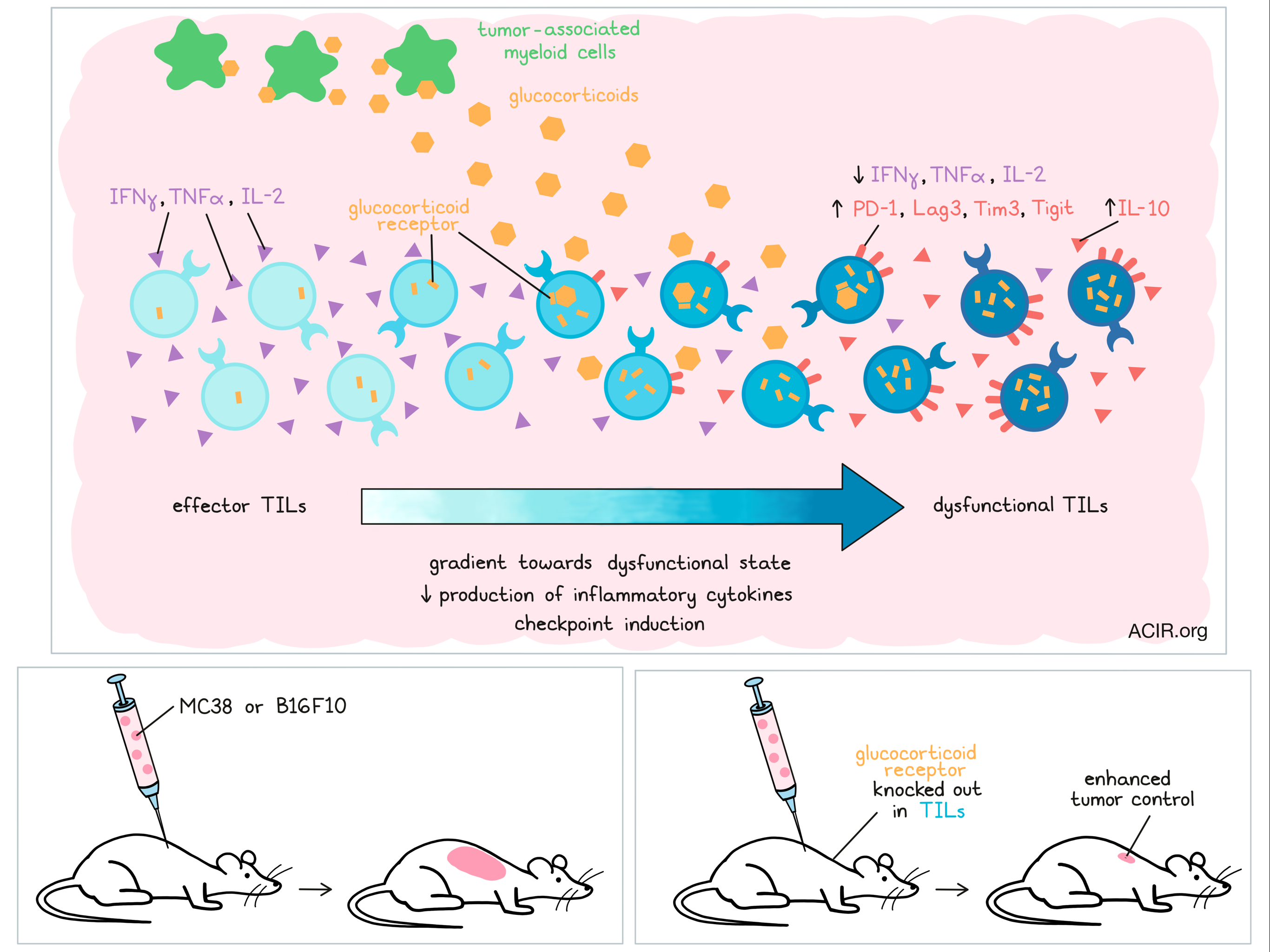
One of the ways the tumor microenvironment (TME) shapes the antitumor immune response is through triggering the presence of exhausted, dysfunctional CD8+ tumor-infiltrating lymphocytes (TILs). Many of the factors contributing to the induction of dysfunctional cells remain largely unknown, but could serve as novel immunotherapy targets. Acharya and Madi et al. recently described their assessment of glucocorticoid signaling as such a mechanism in Immunity.
Glucocorticoids (GCs) are steroid hormones that, once bound to the glucocorticoid receptor (GR) within the cytosol, regulate gene expression. Known for their anti-inflammatory properties, GCs are used to manage inflammation in patients treated with checkpoint inhibitors and to reduce edema in patients with glioblastoma. However, the effects of endogenously produced GCs in the TME remain to be elucidated.
To study this pathway, the researchers first analyzed Nr3c1, the gene encoding the GR, in murine TILs, and found that this gene was mainly expressed by effector (Tim3-PD-1+) and dysfunctional (Tim3+PD-1+) CD8+ TIL. Using MC38 colon cancer and B16F10 melanoma mouse models, they further confirmed a gradient of expression of the GR in CD8+ TIL, increasing from effector to dysfunctional Tim3+PD-1+ T cells. The highest expression of the GR in human colon carcinoma TILs was also found in the Tim3+PD-1+CD8+ T cell subset.
GC expression in B16F10 melanoma CD8+ TILs showed a similar pattern. Cells with low expression of a GC signature were enriched for genes associated with naive T cells, those with medium expression were associated with effector genes, and cells with high expression had high levels of genes related to dysfunctional T cells.
Based on these findings, the authors hypothesized that GC–GR signaling promotes dysfunction characteristics in CD8+ T cells, and evaluated this by repeatedly activating mouse CD8+ T cells in vitro with or without the synthetic GC dexamethasone. Compared to untreated cells, GC-treated cells produced less IL-2, TNFα, and IFNγ, and more IL-10. Furthermore, these cells expressed the genes encoding the checkpoint inhibitors PD-1, Tim3, and Lag3, and in both mouse and human samples, GC increased the frequency of Tim3+PD-1+CD8+ cells.
After confirming the role of GC signaling in differentiating CD8+ T cells into a dysfunctional state, the researchers evaluated what impact the loss of GC signaling had on this process by analyzing TILs from mice with a specific deletion of Nr3c1 in CD8+ cells. These CD8+ T cells did not express the GR and provided better tumor control in both the MC38 and the B16F10 models. While the frequency of TILs was not different between the mice, the TILs with the deletion in Nr3c1 produced more IL-2, TNFα, and IFNγ upon specific and nonspecific stimulation. Additionally, antigen stimulation induced more granzyme B+CD107a+ cells, and the cells with the deletion had reduced expression of the gene encoding TCF-1, a gene known to play a critical role in regulating effector T cell differentiation.
At the intermediate stage of tumor growth, CD8+ T cells with the Nr3c1 deletion were less likely to express, and expressed lower levels of PD-1, Tim3, Lag3, and Tigit. The few Tim3+PD-1+ cells that were present also produced fewer proinflammatory cytokines and had limited cytotoxic capacity. Moving to TCGA data, the authors confirmed a positive correlation between N3RC1 and HAVCR2, PDCD1, LAG3, TIGIT, and IL10 RNA levels in human colon adenocarcinoma.
To assess the mechanisms behind this upregulation of checkpoint receptors, the researchers determined whether the GR directly regulates the expression of checkpoint receptors by analyzing GR-binding peaks in chromatin immunoprecipitation sequencing data of mouse bone marrow-derived macrophages (BMDM). Peaks were found for Havcr2 and Lag3, but not for Pdcd1 and Tigit, likely due to the low expression of these receptors on BMDM. Using luciferase reporter assays, the researchers then tested the effect of GR binding to the cis-regulatory elements in the loci of Havcr2, Pdcd1, Lag3, and Tigit. They added the reporter constructs along with Nr3c1-expressing vectors or empty vectors into 293T cells and cocultured those with GC. In cells expressing the GR, Tim3, PD-1, Lag3, and IL-10 were all potently transactivated. These data suggest that the GR might directly drive the expression of T cell dysfunction genes. Indeed, CD8+ T cells repeatedly stimulated with GC upregulated dysfunction signature genes, including Tim3 and PD-1, while genes associated with Tim3-PD-1-CD8+ effector cells were suppressed.
Acharya and Madi et al. then determined that tumor-associated monocyte-macrophage lineage cells were the predominant local source of glucocorticoids in the TME. These cells expressed the highest levels of Cyp11a1, the enzyme catalyzing the breakdown of cholesterol into pregnenolone, a steroid precursor. Deletion of Cyp11a1 in myeloid cells reduced tumor growth and corticosterone production in MC38 tumors, and the phenotype of CD8+ TILs in this model was similar to those with the Nr3c1 deletions. Other enzymes involved in canonical GC biosynthesis were also present in these myeloid lineage cells, and culturing these cells with Metyrapone, a GC synthesis inhibitor, reduced the production of corticosterone. When Metyrapone was administered intratumorally, tumor growth was inhibited and CD8+ TIL resembled the phenotype of those in the Nr3c1 and Cyp11a1 deletion studies. Together, these data suggest that tumor-associated myeloid cells may trigger the dysfunctional state of TILs by producing GC.
The researchers also found relationships between the GC pathway and outcome and therapy response in humans. Patients with colon carcinoma and stomach adenocarcinoma with low levels of Cyp11a1 in TCGA had a survival benefit. Mice with the Nr3c1 deletion responded better to checkpoint inhibition therapy, and patients with melanoma who had no response to checkpoint inhibitor therapy expressed high levels of the GC and dysfunctional TIL gene signatures in their TILs.
Once further confirmed in human studies, the data presented here could point to improved patient selection for checkpoint therapy and new combination strategies to limit GC signaling, thereby reducing the number of dysfunctional T cells in the tumor microenvironment and increasing immune control of tumors.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first co-author Nandini Acharya and lead author Ana Anderson answered our questions.

What prompted you to tackle this research question?
N.A: Tumor microenvironments play a critical role in shaping immune responses. Understanding factors that promote T cell dysfunction is instrumental in devising novel therapeutic strategies. We analyzed the RNA profiles of the CD8+ tumor-infiltrating lymphocytes (TILs) to get clues about the local signals they receive. Nr3c1, the gene encoding the glucocorticoid receptor, was one of the most highly expressed genes in terminally dysfunctional CD8+ TILs. We were extremely intrigued by this data, as it suggested that glucocorticoid signaling could potentially promote T cell dysfunction. Of note, glucocorticoids are used to treat immune related adverse events (irAEs) following treatment with immune checkpoint blockade in cancer patients. It was important to investigate the significance of glucocorticoid signaling on CD8+ TILs and how this would impact the efficacy of immune checkpoint therapy.
A.A: After years of in-depth study of the factors that work within CD8+ T cells to regulate their functional properties, I came to the realization that this line of research was going to hit a ceiling. I thought to myself that if we really wanted to move the needle in the field of immunotherapy, we needed to understand the signals present in the tumor microenvironment that signal T cells to adopt unfavorable responses. In short, we needed to figure out how to “rewire” the tumor microenvironment. This led us to look at the gene expression profiles of CD8+ T cells from tumors for clues as to the environmental signals that they may be sensing. This is what sparked our investigation into glucocorticoid signaling.
What was the most surprising finding of this study for you?
N.A: Local production of glucocorticoids by the tumor-infiltrating monocytes-macrophage lineage cells was the most surprising finding for me. Our data suggest that the endogenous glucocorticoids are an important immune-suppressive mediator in the tumor microenvironment. We are now investigating the mechanisms that regulate glucocorticoid production from the immune cells in different disease settings.
A.A: What was most surprising to me was the finding that the level of glucocorticoid receptor expression in different CD8+ T cell subsets had different regulatory outcomes. This highlights the complex and cell state-dependent role of glucocorticoid-mediated regulation. Unraveling this is our next experimental challenge.
What was the coolest thing you’ve learned (about) recently outside of work?
N.A: Recently I took up gardening and built a mini indoor garden. Seeing a plant grow makes me feel the real essence of life in the process. I can't really describe the thrill when a flower opens up from its bud and blooms. To me a flower in bloom exudes positive energy, peace and calm. I have several different varieties of orchids, as I simply love them. They last very long and are so exquisitely beautiful. I just learned that some species of orchids can live up to 100 years, and am curious to see how long mine will live!
A.A: I learned about “blue zones”, geographic areas where individuals are known for their longevity, with most individuals living into their 100s. Interestingly, the geographic areas are diverse, but the inhabitants share common lifestyle choices: plant-heavy diets with a lot of legumes, regular physical and social activity, an emphasis on family, and no smoking. I learned that the secret to life is simple: eat what nature provides, keep good company, and keep moving.



