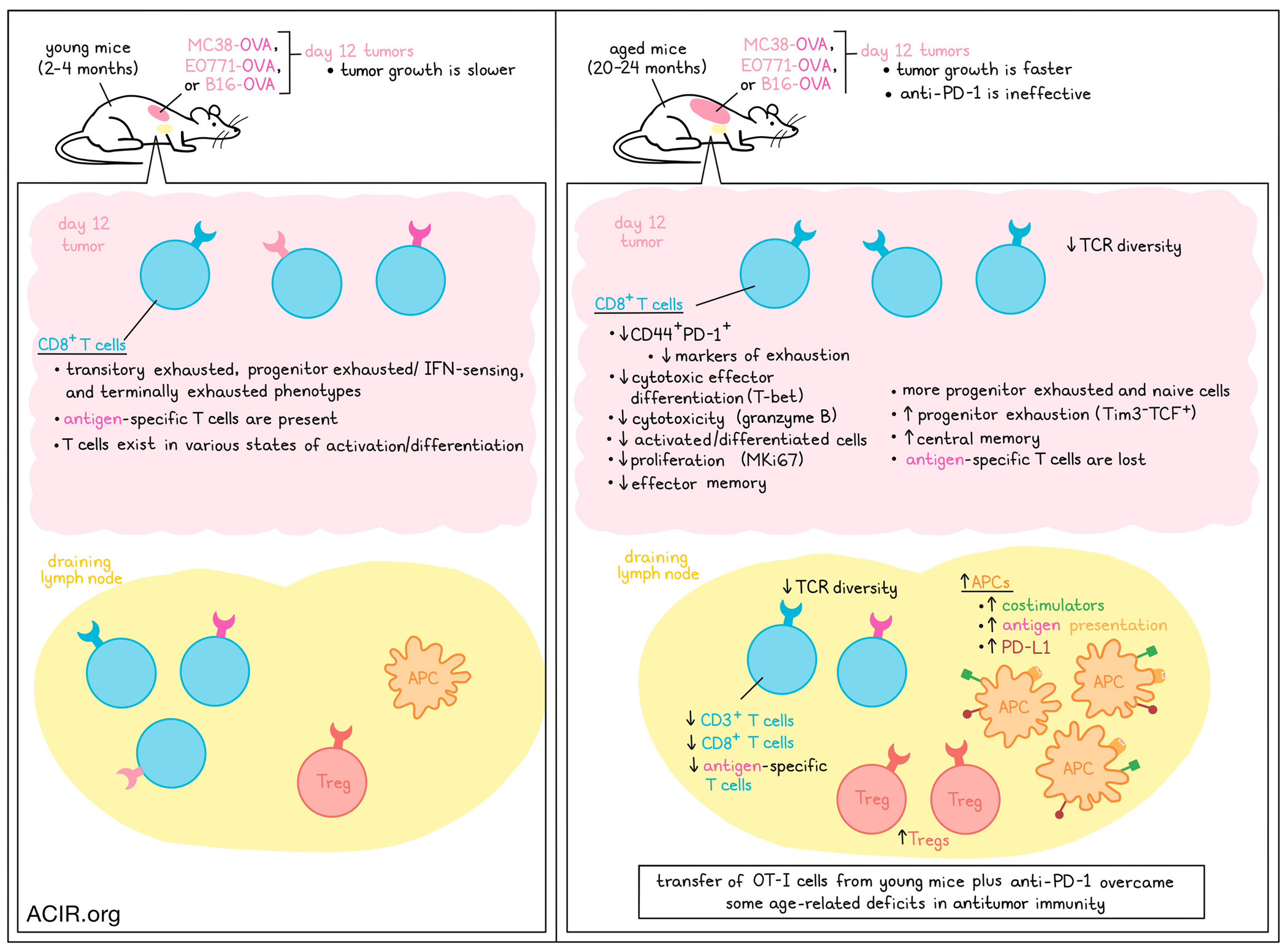
Age-related changes to the immune system contribute to increased cancer progression and resistance to immunotherapy in older patients. Investigating this, Georgiev and Han et al. recently found that tumors grew more rapidly in aged mice due to impaired cytotoxic CD8+ T cell activation and effector differentiation within tumors, and a loss of tumor antigen-specific CD8+ T cells. These findings, along with further investigation into how to overcome these age-related deficiencies, were recently published in Cancer Immunology Research.
To study age-related changes to antitumor immunity, Georgiev and Han et al. evaluated MC38, EO771, and B16 tumors in young adult (2-4 months) and aged (20-24 months) mice. MC38 and EO771 tumors, both of which are immunogenic, grew more rapidly in aged mice, while B16 tumors, which are less immunogenic, grew similarly in young and aged mice. Expression of OVA in MC38, EO771, and B16 tumors enhanced the magnitude of the difference in tumor growth between aged vs. young mice across all models, including in the B16 model, where OVA expression induced a distinction. Depletion of CD8+ T cells accelerated tumor growth in young, but not aged mice, suggesting existing defects in antitumor CD8+ T cells in the aged mouse models.
Investigating the T cell compartment in young versus aged mice bearing OVA-expressing tumors, the researchers found that in aged mice, CD3+ T cells and CD8+ T cells were reduced in tdLNs, but not tumors. Within tumors, a smaller proportion of intratumoral CD8+ T cells in aged mice expressed both CD44 (antigen experience) and PD-1 (activation/exhaustion), and within CD44+PD-1+CD8+ T cells, fewer cells expressed markers associated with exhaustion. TILs in aged mice also expressed reduced markers of cytotoxic effector differentiation (T-bet), cytotoxicity (granzyme B), proliferation (MKi67), and effector memory, and increased markers of progenitor exhaustion (Tim3-TCF+) and central memory, consistent with impaired effector functions and differentiation. This defect was likely not related to impaired priming by APCs, as aged mice actually showed evidence of increased APCs with enhanced antigen presentation and costimulatory functions in tdLNs, though DCs expressed higher levels of PD-L1 and Tregs were increased in these tissues.
Next, the researchers performed scRNAseq and clustering analysis on tumor-infiltrating CD8+ T cells from MC38 tumors. While CD8+ T cells from aged mice fell more into progenitor exhausted and naive clusters, those from young mice fell more into transitory exhausted, progenitor exhausted/IFN sensing, and terminally exhausted clusters. Comparing quiescent and stimulated cells, the researchers noted more differentially expressed genes by age in quiescent cells. Further, trajectory analysis showed that while CD8+ T cells followed a similar differentiation trajectories in young and aged mice, those from young mice were distributed across the range of the trajectory, while those from aged mice were crowded towards the naive end of the pathway, suggesting fewer cells in activated states.
Investigating whether reduced activation was related to reduced antigen-specific CD8+ T cells in aged mice, the researchers found that while SIINFEKL-reactive CD8+ T cells could be readily detected in OVA-expressing tumors on day 12 in young mice, they were essentially absent from those in aged mice at the same time point. In tdLNs, SIINFEKL-reactive CD8+ T cells were detectable in aged mice, but were decreased compared to in young mice. Looking at CD8+ T cells specific for endogenous antigens in B16-OVA (MuLV p15E, gp100, and TRP-2), a similar decline was observed with age.
After determining that these effects could not be attributed to differences in T cell survival or TCRβ expression levels, Georgiev and Han et al. evaluated changes in TCR diversity over time by measuring TCR Vβ chain usage. In young mice, the pattern of Vβ expression was relatively consistent between animals in tissues, but varied between animals in tumors. In aged mice, the central memory pool became dominated by single Vβ chains in spleens and lymph nodes, and variation in naive cells increased between animals in spleens, lymph nodes, and tumors, suggesting reduced TCR diversity in aged mice.
Looking at how the frequencies of CD8+ T cells recognizing SIINFEKL versus TRP-2 varied over time in B16-OVA-bearing mice, the team found that the proportions and absolute numbers of intratumoral antigen-specific CD8+ T cells progressively reduced in tumors with age. This occurred earlier and was more pronounced for SIINFEKL-reactive CD8+ T cells than for TRP-2-reactive T cells. Interestingly, while granzyme B expression and proportions of TIM3+PD-1+ cells remained consistent in SIINFEKL-reactive CD8+ T cells, those features declined with age in TRP-2–reactive CD8+ T cells, suggesting that T cells with different antigen specificities may show distinct kinetics in their age-related declines.
Exploring opportunities to overcome age-related CD8+ T cell deficiencies, the researchers transferred OT-I CD8+ T cells from young mice into young or aged mice prior to B16-OVA tumor implantation. In young mice, this transfer slowed tumor growth, resulting in 25% long-term survival. In aged mice, the treatment initially slowed tumor growth more dramatically than in young mice, but ultimately all tumors escaped immune control, resulting in only a slight extension of survival. OT-I cells were reduced in the tumors of aged mice compared to young mice, but were increased in tdLNs, where a higher proportion of cells expressed MKi67, PD-1, and T-bet.
Next, the researchers evaluated the addition of anti-PD-1 therapy, which on its own, was more effective in young mice than in aged mice, which were unresponsive to treatment. The combination of OT-I and anti-PD-1 therapies was still more potent in young mice than in aged, though it did show some improved antitumor efficacy over OT-I cells alone in aged mice, leading to significantly longer survival and some long-term survivors.
Finally, testing whether age-related changes in CD8+ T cells impact antitumor immunity and responses to immunotherapy in patients, the researchers analyzed data from ~10,000 patients across multiple cancers from TCGA and categorized the patients by a CD8+ T cell infiltration score and age within each cancer type subset. This identified a pattern in which higher diagnosis age was associated with increased lymphocytes, consistent CD8+ T cells, but reduced CD8+ effector T cells. Among younger patients, the progression-free interval (PFI) was found to be significantly longer in individuals with a high CD8+ T cell score, while in older patients, PFIs were not stratified by T cell scores.
Together these results suggest that age-related changes in the immune system, including reduced TCR diversity, loss of tumor antigen-specific T cells, and defects in effector function and differentiation, likely contributing to increased cancer progression and resistance to immunotherapy in older patients. These age-related defects could be overcome with a combination of immunotherapies in mice. These results also support the use of aged mice for modeling cancer research in older patients, who make up the largest portions of most treatment populations.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, co-lead author Alison E. Ringel answered our questions.

What was the most surprising finding of this study for you?
We were surprised to uncover the extent to which antigen-specific killer T cells, which are important for recognizing new threats like tumors, decline with advanced age! This is a mechanism that may contribute to the rise in cancer incidence in later stages of life.
What is the outlook?
On one hand, our work suggests that aging may shift the features of tumors that are recognized by T cells in older individuals. Going forward, we need to understand more about how this affects tumor control by the immune system and if this influences how patients respond to immunotherapy. From a research-focused standpoint, we need to make sure that we accurately account for this decline in tumor-specific T cells when using mice as a preclinical model for human aging. This is important as more and more research focuses on the intersection of aging and antitumor immunity.
What was the coolest thing you’ve learned (about) recently outside of work?
My family recently traveled to Colorado for vacation. We visited an old gold mine with my two kids, where we learned that there’s still a lot of gold left in the mountains in Colorado, but it costs as much money to get it out as it’s worth!



