Gene expression editing in tumor cells results in evasion of immunosurveillance, but the exact edited genes and their relationship to the immune response have not been studied in detail. Using a genetically engineered breast cancer model, Zhang, Naderi Yeganeh, et al. identified edited genes related to the immune response, and evaluated a therapeutic approach to overcome these effects. Their findings were recently published in Nature Immunology.
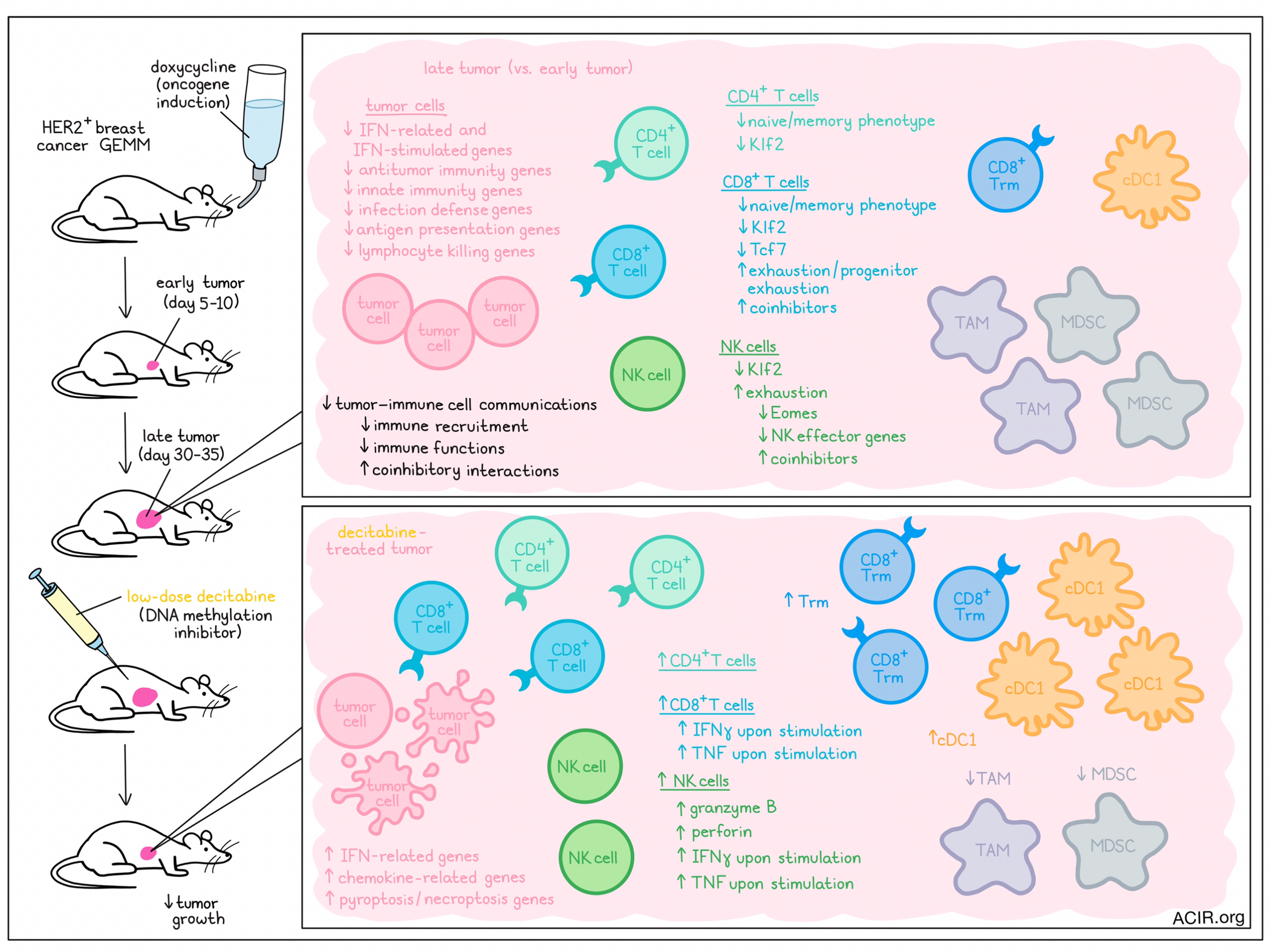
Compared to most murine tumor models, genetically engineered mouse model (GEMM) tumors grow naturally in the tissue, more closely resembling human tumor growth. To evaluate genetic changes during tumor progression, the researchers assessed an aggressive breast cancer GEMM in which mice develop metastatic HER2+ breast tumors that are resistant to immune checkpoint blockade. To detect early changes during tumorigenesis, tumors harvested at days 5-10 (early) or days 30-35 (late) after oncogene induction were assessed by scRNAseq.
Clustering analysis was performed, and cells in clusters resembling tumor cells were pooled to determine the differentially expressed genes between early and late tumor cells. In late tumor cells, 229 genes were upregulated, and 128 genes were downregulated. Some of the downregulated genes were related to interferons and interferon-stimulated genes. Gene ontology (GO) enrichment analysis of downregulated tumor genes revealed 179 significant mostly immune-associated GO terms. Most overrepresented terms among downregulated genes were involved in innate immunity and infection defense, as well as adaptive immune pathways involved in antigen processing and presentation and lymphocyte killing. Upregulated genes were related to oncogenic signaling, cell proliferation and survival, metabolism, stress responses, or immune inhibition. However, enrichment terms of downregulated genes had much lower p-values than those of upregulated genes, suggesting tumor editing predominantly results in suppression of tumor immunity, rather than in gene alterations that change tumoral behavior.
Since pyroptosis and necroptosis can cause immunogenic cell death, the researchers looked into related pathways, but did not find differentially expressed genes in early vs. late tumors, suggesting no significant editing of these genes.
Moving on to the tumor-infiltrating immune cells, the scRNAseq data were used to compare T and NK cell characteristics of early and late tumors. This revealed that the proportion of naive CD4+ and CD8+ T cells was significantly reduced, while progenitor exhausted and exhausted CD8+ T cells increased in the late tumors. There were also overall changes in the CD4+, CD8+, and NK cell populations, with a reduction in the transcription factor Klf2, which controls naive lymphocyte homing to secondary lymphoid tissue. Late tumor CD4+ and CD8+ T cells had reduced expression of naive and memory cell marker transcripts, and CD8+ T cells downregulated Tcf7, while upregulating many coinhibitory receptor genes. NK cells also showed characteristics of exhaustion, with reduced Eomes and NK effector genes, and increased coinhibitory receptor gene expression.
The researchers then assessed the inferred cell–cell interactions in early and late tumors based on scRNAseq data, focused on changes in ligand-receptor gene coexpression in all cells. Over 107 significant cellular communication pathways were active in early or late tumors, with 44 being more active in early samples, and 29 having more activity in late samples. Additionally, 13 pathways active in early tumors were inactive in late tumors, and these included innate and adaptive immune pathways.
Zooming in on cell communication pathways between NK or CD8+ resident memory T (Trm) cells and tumor cells, multiple ligand–receptor interactions were detected in early tumors that disappeared in late tumors. These included interactions related to immune cell recruitment and function. Further, there was an increase in coinhibitory signaling interactions between CD8+ T cells and tumor cells in late tumors, which was mostly due to increased expression of coinhibitory receptors on CD8+ T cells.
Since DNMT promoter methylation is one mechanism of repression of gene expression, Zhang, Naderi Yeganeh, et al. assessed whether treatment with the DNA methylation inhibitor decitabine could improve the immunogenicity of late tumors. Mice with late GEMM tumors were treated with decitabine, which reduced tumor growth. Furthermore, tumors had increased levels of CD4+, CD8+, and NK cells, with a high increase in Trm cells and cDC1s, while levels of immunosuppressive myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages decreased. NK cells expressed more granzyme B and perforin, and more CD8+ T and NK cells from these tumors produced IFNγ and TNF after ex vivo stimulation. In treated tumors, there was an increase in genes related to necroptosis, pyroptosis, and genes related to IFN pathway and chemokines.
Next, the 4T1 triple-negative breast tumor model was treated with three doses of decitabine. In this model, therapy also improved antitumor immunity and delayed tumor growth. There was an increase in total CD8+ T cells, Trm cells, and cDC1s, and a decrease in MDSCs.
In vitro, the effects of low-dose decitabine treatment on 4T1 was assessed, with a focus on 18 type I IFN and inflammation pathway mRNAs. Low doses did not affect cell viability, but upregulated most of the genes assessed. Treatment resulted in the upregulation of 2,622 genes and downregulation of 717 genes, and there was significant overlap in genes upregulated by treatment and those downregulated in late GEMM tumors. Most of these overlapping genes were involved in IFN/cytokine signaling, antigen presentation, or cell killing.
To further assess the role of the type I IFN pathway, the researchers assessed the effects of decitabine in Ifnar1-/- mice. Since these mice are only available on C57/Bl6 background, the B16F10 melanoma model was used. Tumor growth in both WT and Ifnar1-/- mice were both inhibited by decitabine treatment, but the tumors overall grew much faster in Ifnar1-/- mice, independent of decitabine. Immune infiltration was comparable between treated WT and Ifnar1-/- mice. In WT mice bearing B16F10 tumors, decitabine induced apoptosis, necroptosis, and pyroptosis, suggesting it induced inflammatory cell death. Further, treatment impacted type I IFNs, resulting in tumor growth suppression and immune stimulation.
In conclusion, this study shows the effects of tumor editing of gene expression on immune evasion, and how the immune suppressive effects of editing might be overcome by inhibition of DNA methylation. This may open the door to more treatments aimed at targeting immune-cold tumors, with the aim of improving immunotherapy responses.
Written by Maartje Wouters, image by Lauren Hitchings



