The ability to induce de novo, potent, and truly tumor-specific T cell responses as shown in two independent studies with different vaccine formats by Ott & Hu et al. (Ott&Hu) and Sahin et al. (Sahin) has the potential to transform cancer vaccination. T cells directed against neoantigens, which arise from non-silent somatic mutations that are unique to each patient’s tumor, are now known to be central to the mechanisms of action of modern immunotherapies. However, to capitalize on the encouraging biology of neoantigens, it is necessary to overcome the hurdles of preparing a personalized vaccine, a challenge now met by these two studies.
To generate the vaccine, both groups used whole exome and RNA sequencing to identify and validate non-synonomous single nucleotide variant mutations and predicted neoepitopes capable of binding to the patient’s individual MHC class I molecules to activate CD8+ T cells. As points of difference, Ott&Hu also included out-of-frame insertion and deletion mutations, and Sahin predicted MHC class II binding neoepitopes to stimulate CD4+ T cells in two patients.
Ott&Hu vaccinated patients with up to 20 long peptides, each covering a specific mutation and potentially capturing MHC class II neoepitopes, together with the TLR3 agonist poly-ICLC as adjuvant, subcutaneously. Sahin injected patients with two synthetic RNA molecules, each encoding five linker-connected long peptides, directly into the lymph node. By targeting multiple neoepitopes simultaneously, both studies address tumor heterogeneity and immunologic escape through antigen loss.
Both studies enrolled high-risk, stage III and IV melanoma patients. Ott&Hu treated six patients, two of whom received IFN-α as prior systemic therapy, and all of whom had no evidence of disease following surgery. Sahin treated thirteen patients, many of whom had received one or more prior systemic therapies, and 5 of whom had evidence of disease at the initiation of vaccination. Nine of the 13 patients in the Sahin study received an RNA vaccine against the tumor-associated self-antigens (TAA) NY-ESO-1 and/or tyrosinase prior to neoantigen vaccination.
Vaccination induced CD4+ and CD8+ T cell responses against multiple neoepitopes in all of the treated patients. Remarkably, T cell responses were detected against 60% of the vaccinating neoepitopes. While Sahin detected weak pre-existing T cell responses for a third of the vaccine-induced responses, Ott&Hu observed only de novo responses. In both trials, a fifth of the vaccinating neoepitopes stimulated T cell responses which could be detected directly ex vivo, while the others were seen after one round of in vitro stimulation. Proportionally, more CD4+ than CD8+ neoepitope-specific T cells were induced. In each trial, two thirds of the epitopes that elicited specific CD8+ T cells also induced CD4+ T cells. T cells were found to be polyfunctional, secreting multiple effector cytokines, and effectively distinguished between wild-type and single-amino-acid-mutated antigens. Of note, Sahin reported significantly stronger T cell responses against neoepitopes compared to the shared TAAs.
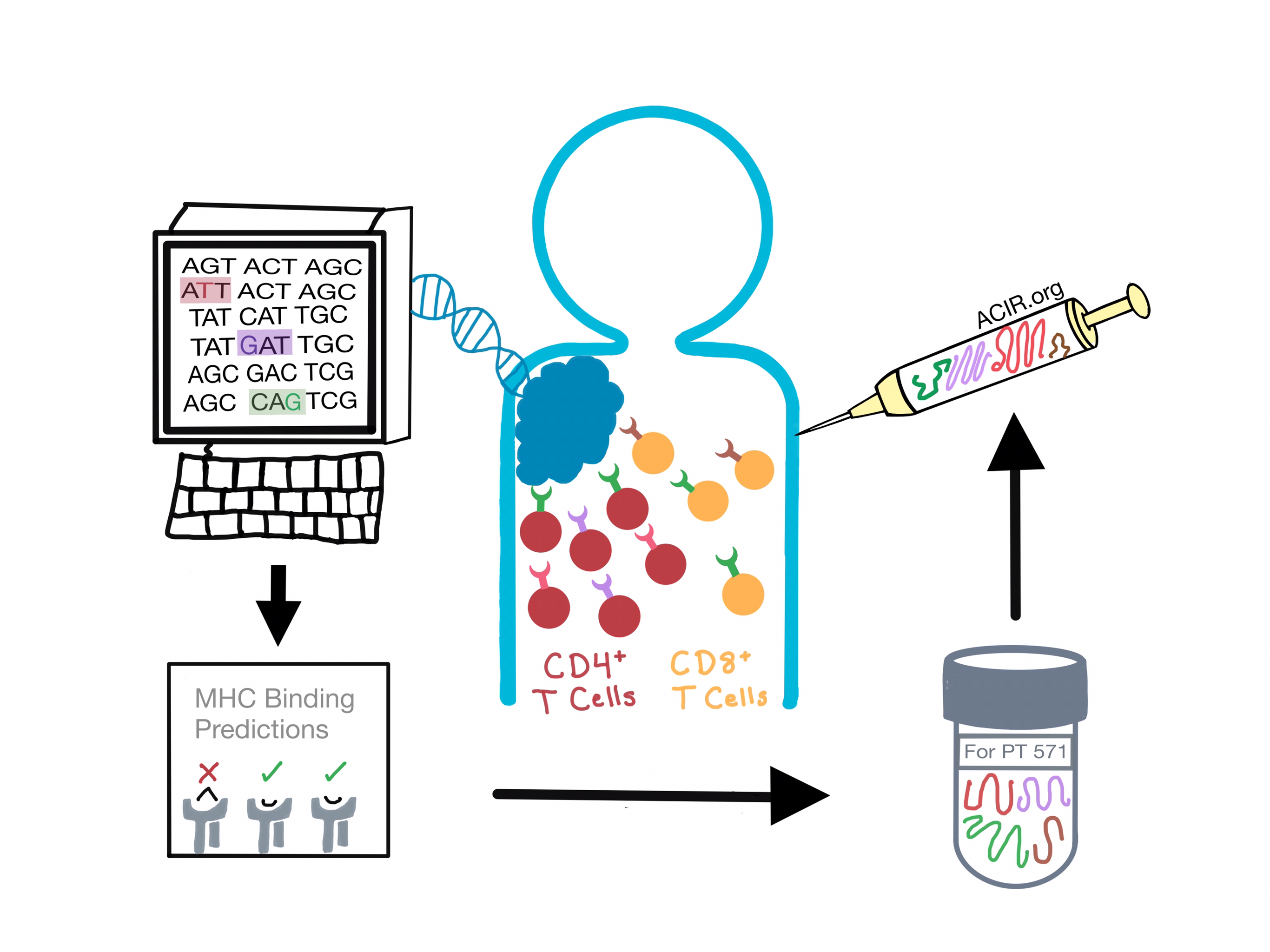
Ott&Hu found that 73% of the neoantigen-reactive CD4+ cells and 100% of the CD8+ cells recognize endogenously processed and presented neoantigens on transfected B cells. Furthermore, Ott&Hu verified MHC class I and II expression on originally resected metastatic tumors, and both studies showed that neoepitope-specific CD4+ and CD8+ T cells were reactive against the patient’s own melanoma. Finally, Sahin confirmed the infiltration of vaccine-induced T cells in the cognate tumor, suggesting that neoantigen-specific T cells are effective in vivo.
The historic rate of recurrence for high-risk melanoma two years after surgery is ~50%, but the majority of vaccinated patients remained recurrence free for the follow up period of 20-32 months (4 patients of Ott&Hu) and 12-23 months (10 patients of Sahin). In each study, two patients who experienced recurrence were transitioned onto anti-PD-1 checkpoint blockade therapy. Three of these four patients experienced complete and durable tumor regression. Immune analysis 9 to 12 months later demonstrated that the repertoire of vaccine-induced T cells persisted and that additional neoantigen responses could be detected. One of the anti-PD-1 treated patients died due to outgrowth of melanoma with defective MHC class I presentation (β2-microglobulin loss).
The demonstrated feasibility of preparing a truly personalized vaccine based on each patient’s unique tumor along with the highly specific, potent immunological responses that were induced in these two early clinical trials open up a whole new approach to cancer immunotherapy.
by Dominique Stavis and Ute Burkhardt



