
Last week, the ACIR team attended the CIMT Annual Meeting 2022 in Mainz, Germany. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Personalized cancer immunotherapy
Alena Gros Vidal
Hajer Fritah Guiren
Jeroen van Bergen
CAR T cell therapies
Hideho Okada
Andreas Mackensen
Luca Gattinoni
Antigen selection
Celina Tretter
Megan Burger
Andrew Sewell
Jonas Ibn-Salem
Cancer vaccination strategies
Mads Hald Andersen
Matthias Eyrich
Dissection of immune responses
Jolanda de Vries
Douglas Hanahan
Daniela Thommen
Targeting the tumor immune microenvironment
Michele de Palma
Martina Seiffert
Jӧrg Wischhusen
Assunta Cirella
Personalized cancer immunotherapy
Refining the identification of personalized tumor-reactive lymphocytes- Alena Gros Vidal - Vall d'Hebron Institute of Oncology (VHIO), Barcelona, Spain.
In order to be able to better understand natural or therapy-induced antitumor immunity, and develop more effective immunotherapies, Alena Gros Vidal set out to identify biomarkers that can help guide reliable identification of tumor-reactive lymphocytes. Gros Vidal’s early studies have shown CD8+PD1+LAG3+ TILs to be enriched in tumor-reactive T cells. Other publications have described CD103+, PD-1+, TIM3+LAG3+, 4-1BB+, CD103+CD39+, CD39+, PD-1hiCD39+, PD-1hiICOS+ as markers that are preferentially expressed on tumor-reactive CD8+ or CD4+ TILs. However, bystander T cells, changes in frequency of T cell clones upon TIL culture, T cell reactivity and differentiation, and availability of an autologous T cell line have complicated the quest for reliable biomarkers. Some of these hurdles can be overcome by leveraging single-cell transcriptomic data to analyze the functional state and phenotype of tumor- or neoantigen-specific TILs. Gros Vidal and team prospectively studied the TIL phenotype in a cohort of 47 patients with primary endometrial cancer (EC), a tumor that is characterized by 4 mutational subtypes (POLE, MSI, Copy Number Low [CNL], and Copy Number High [CNH]). POLE-mutated EC tumors display a higher frequency of CD3+ TILs compared to the other mutational EC subtypes, but this was not associated with a positive prognosis. Zooming in further, autologous tumor-reactive T cells were enriched in a CD8+ subset that is PD-1hiCD39+CD103+CXCL13+ and a CD4+ subset that is PD-1hiCD39-, and typically CXCL13+. Proportions of both subsets correlated with improved survival. Even though it was at much lower frequency compared to TIL, Gros Vidal also found CD4+ and CD8+ circulating tumor-reactive T cells to be enriched in the PD-1hi population.To refine the identification of circulating neoantigen-reactive lymphocytes, Gros Vidal and team first sorted blood CD4+ and CD8+ T cells from patients with various cancer types based on PD-1 and other markers, and took tumor biopsies for the identification of neoantigens by whole-exome sequencing. Autologous APCs pulsed with mutated peptides or electroporated with tandem minigenes were then used for coculture with the sorted T cells. Sixteen neoantigen-specific and 3 cancer germline-specific T cell reactivities were detected in 8 out of 10 patients. Of all markers and marker combinations studied, PD-1hi and CD39 co-expression was consistently found to most reproducibly enrich for CD8+ and CD4+ neoantigen-specific lymphocytes in the blood, although the CD4+ population showed slightly more diversity of markers. Multiple independent TCRs could be detected for a single neoantigen. The use of biomarkers to select and enrich for tumor-reactive lymphocytes from the tumor and blood holds promise to accelerate the development of personalized T cell therapies.
Personalized cancer vaccination strategies: whole tumor-based vaccines versus neoepitope-based vaccines- Hajer Fritah Guiren - Ludwig Institute for Cancer Research, University of Lausanne Lausanne, Switzerland.
The presence of intratumoral T cells is clearly linked with long-term patient benefit, but what are the features that define useful epitopes, and how could they be applied to the selection of epitopes for neoantigen cancer vaccines? To address these questions Hajer Fritah Guiren began by comparing various methods of neoepitope prioritization with the epitope-agnostic whole tumor cell lysate (WTC) vaccine, which has demonstrated good immunogenicity and clinical benefit in ovarian cancer. After showing in two murine tumor models (LLC1 and B16F10) that dendritic cell-delivered WTC vaccines were effective in therapeutic tumor control, Fritah Guiren began dissecting the features and impact of the sequence-detected neoepitopes. Neoepitopes were ranked by in silico binding prediction, and HLA in vitro binding affinities were determined biochemically. Furthermore, the in vivo immunogenicity of each peptide known to bind in vitro was tested. Interestingly, there was no correlation between either in silico-predicted or in vitro-determined binding rank and immunogenicity, indicating the lack of utility of such selection approaches. Moreover, the top 5 in silico-determined peptides together were unable to control tumor growth as well as WTC, while the top 5 in vitro binding-determined or immunogenic peptides were able to control tumors comparably to WTC. Drilling down to individual epitopes, Fritah Guiren identified epitopes that were immunogenic following WTC vaccination. Of the top 5 neoepitopes identified by each approach, only two were immunogenic with the WTC vaccine, and of these, the neoepitope identified by both in vitro binding and immunogenicity was able to control tumor, while the epitope identified in silico and by immunogenicity was unable to control tumor, setting a high bar for the features of useful neoepitopes. Fritah Guiren then turned to analyzing human samples following either of two DC-based vaccine regimens (WTC in ovarian cancer [OC] or predicted neo-peptides in non-small cell lung cancer [NSCLC]). In the OC study, there was no correlation between in silico score and immunogenicity, but there was clear enrichment of in vitro binding-identified epitopes among those that were immunogenic. Further, in the NSCLC study, in vitro binding more strongly correlated with immunogenicity than in silico prediction. Fritah Guiren closed by suggesting priming with a WTC vaccine and boosting with a defined epitope vaccine based on in vitro binding determination.
Personalized synthetic polyepitope DNA cancer vaccines including a novel pyroptotic adjuvant synergize with checkpoint inhibitor therapy to generate effective anti-tumor T cell immunity- Jeroen van Bergen - Immunetune, Leiden, The Netherlands.
To develop more effective and time- and cost-conserving neoantigen cancer vaccines, Jeroen van Bergen described two novel approaches to DNA-based vaccines. Building on standard sequencing, mutation calling, and construction of a polyepitopic vaccine, van Bergen described a novel DNA-encoded adjuvant, PyroVantTM, which encodes a constitutively active caspase 1 inflammasome stimulator. Constitutive inflammasome stimulation mimics a pathogenic infection, and importantly, active caspase 1 stimulates Gasdermin D production, which creates holes in the cell membrane, releasing DAMPs, IL-1β, IL-18, and antigen, which activate and load antigen-presenting cells. The constitutively active form of caspase 1 was constructed by eliminating the CARD domain (involved in recruitment of other molecules in the pathway) and swapping the order of the catalytically active P20 and P10 domains. After validating in vitro that transfection with the PyroVantTM construct, together with pro-IL-1β, led to IL-1β release, and with Gasdermin D, led to LDH release and cell death, van Bergen conducted an in vivo vaccination study with MC38 neoepitopes and OVA (with CD4 helper epitopes). When PyroVantTM was included, there was an ~8x increase in antigen-specific cells a week after vaccination, with typical contraction by 4 weeks to levels comparable to vaccination with antigen alone. Using B16F10 epitopes and a similar single vaccination, challenge with B16F10 tumors at that day-28 timepoint led to significantly better prophylactic tumor control with PyroVantTM. Therapeutic tumor control studies with MC38 or B16F10 showed that PyroVantTM synergized with anti-PD-1 or agonistic 4-1BB therapy more effectively than antigens alone, significantly increasing the fraction of long-term survivors. The safety of PyroVantTM has been shown in animal safety studies. Finally, van Bergen described a GMP-compatible synthetic DNA production approach using enzymatic synthesis.
CAR T cell therapies
Novel "prime-and boost" concept of T cell therapy using the synNotch circuit to overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma- Hideho Okada - University of California, San Francisco, USA.
To succeed in patients with solid cancer, including glioblastoma (GBM), CAR T cell approaches need to overcome the challenges of on-target/off-tumor toxicities, intratumoral heterogeneity of tumor-expressed antigens, and lack of persistence of T cells due to exhaustion and the effects of tonic signaling. Since there are no perfect antigens for immunotherapy of GBM, Hideho Okada decided to go with two imperfect ones. Okada and team created a "prime and kill" circuit by combining synthetic Notch receptors (SynNotch) that recognize specific priming antigens – which are glioma or brain-specific, but not expressed on all tumor cells – with a CAR that targets glioma-associated antigens, which are more uniformly expressed on tumor cells, but are not tumor-specific. Local recognition of the priming antigen induces expression of the CAR to target the “kill” antigens, leading to local killing of tumor cells, while sparing normal tissue that only expresses the glioma-associated antigen, and not the priming antigen. EGFRvIII-triggered synNotch-CAR T cells showed tumor region-restricted priming and killing, an improved naive/stem-like memory phenotype, enhanced persistence in vivo, and reduced exhaustion. Testing different CNS-specific antigens, Okada identified Brevican (BCAN) as the antigen that enabled the most specific and robust priming of the circuit. The BCAN synNotch was combined with a tandem CAR that simultaneously targets EphA2 or IL-13Rɑ2, to generate B-SYNC CAR T cells. A single intravenous infusion of B-SYNC T cells led to sustained complete remission of intracranial patient-derived glioblastoma (GBM6) xenografts without affecting the growth of BCAN-knockout GBM6 tumors in the periphery, confirming the restricted CAR expression in the CNS. B-SYNC efficiently homed to the GBM site and persisted in the brain at day 45 following i.v. infusion. A phase I clinical trial of B-SYNC T cells is in preparation.
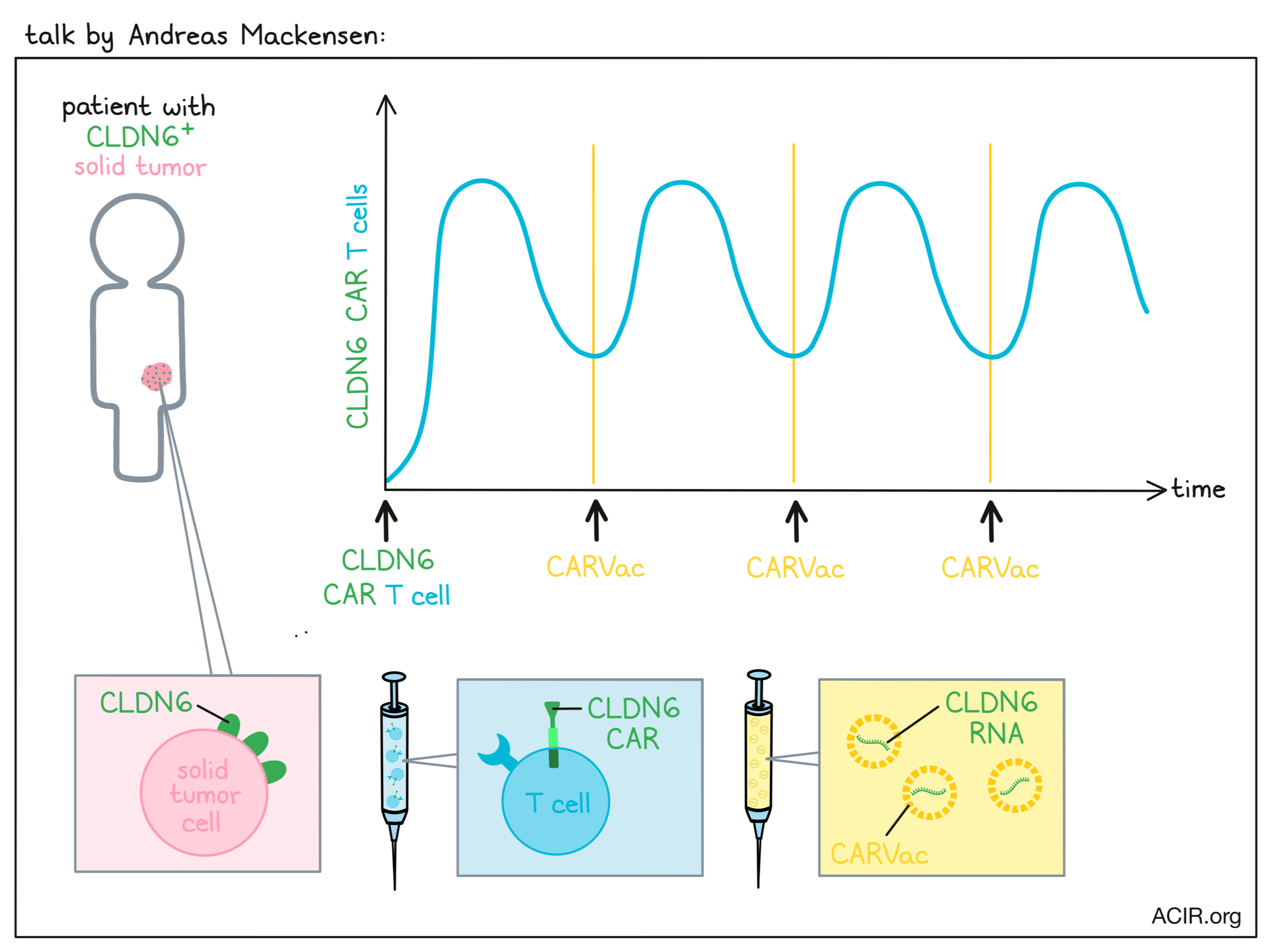
A phase I/II trial to evaluate safety and efficacy of CLDN6 CAR-T and CARVac- based in vivo expansion to improve treatment of patients with CLDN6+ advanced solid tumors- Andreas Mackensen - University Hospital Erlangen, Erlangen, Germany.
Andreas Mackensen discussed a recent clinical trial evaluating the safety and efficacy of Claudin-6 (CLDN6)-targeted CAR T cells in combination with a CARVac – a lipid nanoparticle vaccine loaded with CLDN6 mRNA to amplify CLDN6-CAR T cells. CLDN6 is expressed during development and re-expressed in a number of solid tumors, including ovarian and testicular cancers, making it a strong target with limited on-target off-tumor toxicity. Patients in this trial, who had a variety of tumor types and were heavily pre-treated, were treated with one of two doses of CAR T cells with or without CARVac, which was started at day 4 and was given in 3-week intervals. Two dose-limiting toxicities were prolonged pancytopenia after lymphodepletion and Hemophagocytic lymphohistiocytosis (HLH) before start of CARVac; both were manageable with interventions. Adverse events above grade 3 were also prevalent, but mostly associated with lymphodepletion. A new arm of the trial was opened for patients who had previously undergone high-dose chemotherapy to have a lower lymphodepleting regimen to avoid potential risks. Looking next at CAR T cell kinetics, the researchers found that CAR T cell engraftment was successful in all patients, with persistence observed mainly in responding patients. The addition of CARVac did appear to support T cell engraftment. Evaluating efficacy in the 14/16 evaluable patients, the overall response rate (ORR) was 43%, and the disease control rate (DCR; stable disease or better) was 86%. Patients being treated at dose level 2 plus CARVac had a 75% ORR and a 100% DCR. At the first evaluation, 6 patients showed partial responses, and one patient with testicular cancer had their partial response deepen into a complete response at a later time point. Interestingly, this patient’s T cells were mainly of a TEMRA phenotype, and CAR T cells were still detectable 150 days after the infusion in this patient. At both dose levels, the addition of CARVac contributed to improved responses, and in at least some patients, induced re-expansion of CAR T cells upon administration. Interestingly, CARVac also appeared to upregulate cytokine receptors, including the IL-2 and IL-7 receptors, on CD4+ CAR T cells, which could enhance T cell proliferation, activation, survival, and homeostasis. While early, these results are promising for the use of CAR T cells, bolstered by CARVac, against solid tumor targets.
CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies- Luca Gattinoni - Leibniz Institute for Immunotherapy (LIT), Regensburg, Germany.
Luca Gattinoni discussed a strategy for improving CAR T cell therapy by improving the quality of the T cells themselves. According to Gattinoni, not all T cells are created equally, and different phenotypes and functions may serve better in adoptive cell transfer, including for CAR T cells. For example, memory T cells might be favorable due to the fact that they readily expand and persist long-term, and are less prone to exhaustion or cell death compared to their effector counterparts. Particularly, stem cell memory T cells (TSCM) appeared promising to Gattinoni and team. To select for these cells, the researchers performed serial positive selection for enrichment for naive CD8+ T cells, then used flow cytometry to select for CD8+CD62L+CD45RA+ cells. Compared to products made from unselected cell subtypes, memory subtypes, or memory mixed with naive subtypes, these naive TSCM cells showed the best responses. For CAR T cell manufacturing, T cells could be selected, activated, and transduced with the relevant CAR. Compared to standard CAR T cell products, which are enriched for effector memory phenotypes and have a low frequency of naive-like cells, TSCM CAR T cells are more homogeneous, have gene expression profiles comparable to naturally occurring TSCM cells, and have metabolic profiles of low glycolysis, which is associated with response. Further, in an acute lymphoblastic leukemia mouse model, this selected cell product mediated long-lasting responses and prevented relapse better than the standard product. When tested in a phase I clinical trial, allogeneic CD19 CAR-modified TSCM cells were used to treat patients with B cell malignancies that were refractory to allogeneic hematopoietic stem cell transplant (alloHSCT). Despite 14/20 treated patients having a history of GVHD after alloHSCT, no GVHD was observed in response to CAR T cell therapy. Further, the CD19 CAR TSCM cells were more effective at lower doses than the standard products. Looking at the kinetics of these cells, it appeared that they had a delayed, but higher peak expansion, with lower production of inflammatory cytokines, and reduced inflammatory responses (as measured by fever). Use of integration site analysis to analyze clonal expansion in circulation revealed distinct waves of clonal expansion. No evidence of aberrant clonal behavior was observed, and CAR T cell clones that persisted were preferentially derived from TSCM cells that did not participate in the early expansion phase. Additionally, resistance to treatment was largely due to antigen escape, rather than poor persistence, and severe CRS was found to be driven primarily by tumor burden.
Antigen selection
Integrated multi-variate genomic, immunopeptidomic and functional analyses reveal RNA as a superior source for entity-agnostic neoantigen identification correlating with T-cell infiltration in the tumor- Celina Tretter - German Cancer Consortium of Translational Cancer Research (DKTK) and German Cancer Research Center (DKFZ), Heidelberg, Germany.
Aiming to improve a bottleneck in personal neoantigen vaccination – the optimal selection of epitopes to include – Celina Tretter used a cohort of 32 patients with primary or metastatic cancers of multiple histologies to conduct genomics and immunopeptidomics to identify epitopes, followed by analysis of TILs for neoantigen-specific T cells. Rather than using only paired normal and tumor whole-exome DNA sequencing to identify mutations, Tretter focused on the RNA sequencing data and could identify many more (>10x) single-nucleotide variants by RNA sequencing. A fraction of these RNA variants (~10%) were shared across multiple patients, including genes associated with cancer and cancer testis antigen (CTA)/tumor-associated antigens (TAAs). Immunopeptidomics on tumors also revealed that substantially more neoantigen peptides were detectable from the RNA-identified variants, and that many of these were shared across patients. Using patient and HLA-matched allogeneic TILs and PBMCs in a functional (ELISPOT) assay, 23 of 79 tested shared neoantigen candidates (a total of 91 were identified) were demonstrated to be spontaneously immunogenic and found to be independent of tumor entity. Most of these were derived from RNA sequencing data, and the mutation signature showed a predominance of A-to-I editing as the source of the neoepitopes; thus, these would not be detectable by DNA sequencing. Analysis of T cell infiltration demonstrated that there was a strong correlation between the presence of these immunogenic variants and infiltration of CD8+ T cells, including those with upregulated inhibitory receptors, suggesting that the pipeline revealed highly relevant and shared neoantigens.
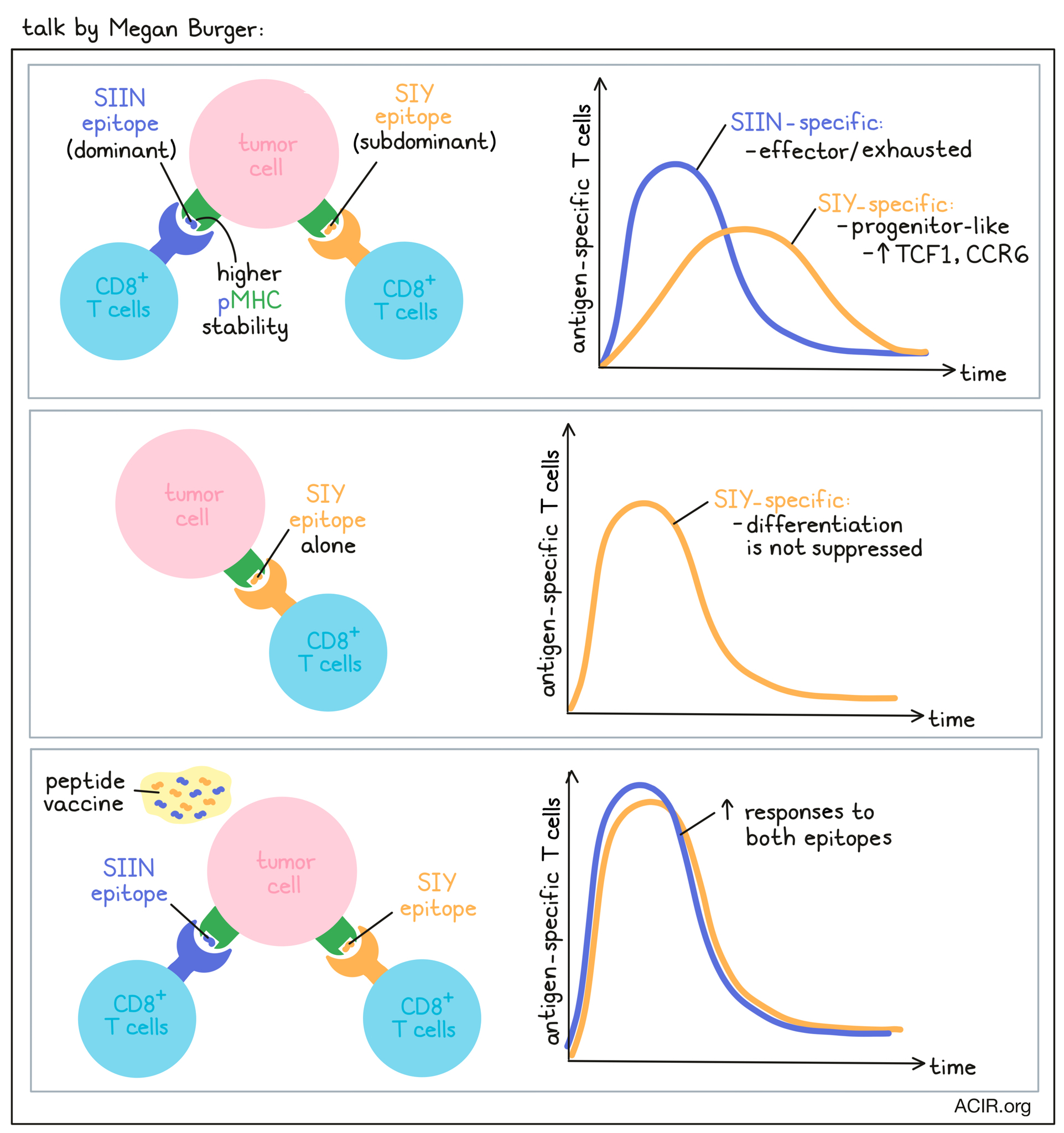
Antigen dominance hierarchies shape anti-tumor T-cell phenotypes and immunotherapy response- Megan Burger - Koch Institute at MIT, Cambridge, USA.
To unravel the factors that regulate tumor-specific T cell immunity, Megan Burger used the conditional KP lung cancer model from the Tyler Jacks lab, in which tumors concurrently express the OVA CD8+ T cell epitope SIINFEKL (SIIN) and the synthetic CD8+ T cell epitope SIYRYYGL (SIY). At early time points (5-8 weeks post tumor initiation), T cells were robustly recruited to tumors, but this was followed by a rapid decline in the CD8+ T cell response and loss of tumor control while tumor antigen expression was maintained. Burger found that T cell responses to the SIIN and the SIY peptides differed considerably. SIIN-specific T cells dominated the CD8+ T cell response, but then declined, while subdominant SIY-specific T cells persisted better over time, with both T cell populations eventually reaching comparable levels. Over time, both T cell populations declined in proliferation and expressed more inhibitory receptors. SIY-specific cells showed delayed kinetics of dysfunction, higher expression of IL-7R and memory markers, and limited effector function compared to SIIN-specific T cells. Phenotypically, the SIIN-specific T cells had an exhausted signature, while SIY-specific T cells displayed a less differentiated TCF1+ progenitor phenotype. When Burger expressed the antigens individually in the tumor to study whether the observed effects were due to the concurrent expression of both neoantigens, an antigen dominance hierarchy emerged, in which the differentiation of SIY-specific T cells was suppressed in the presence of the SIIN epitope. A low-pMHC-stability mutant of SIIN confirmed that pMHC stability, and not affinity, was a key determinant of antigen dominance, with the higher stability epitope resulting in a greater magnitude of response and the lower stability epitope being enriched for the TCF1+ progenitor cell phenotype. Subdominant TCF1+ T cell responses were enriched for a dysfunctional subset expressing CCR6, a chemokine associated with Th17 cells, and these CCR6+TCF1+ T cells were associated with poor response to ICB therapy in the mice. CCR6+TCF1+ T cells could also be found across human cancers. While CCR6+TCF1+ T cells could not predict response to ICB therapy, CCR6-TCF1+ cells were associated with response to anti-PD-1 and/or anti-CTLA-4 therapy. Therapeutic vaccination with long SIY and SIIN peptides and cyclic diGMP substantially improved responses to both epitopes. Notably, while the subdominant SIY response was still enriched for TCF1+ progenitor cells, the TCF1+CCR6+ population was almost eliminated. Burger showed that antigen dominance hierarchies can form in tumors and restrict the breadth and functionality of the antitumor T cell response, and that therapeutic vaccination can break these hierarchies and help to better engage the full breadth of the T cell response.
New modes of T-cell recognition and novel broadly-expressed T-cell epitopes by dissection of cancer immunotherapy success- Andrew Sewell - Cardiff University, Cardiff, UK.
Asking the novel, but in retrospect, obvious and highly reasonable question, “what do successful T cells recognize?”, Andrew Sewell assembled a cohort of patients cured by TIL therapy or checkpoint inhibition, and other extreme survivors. His talk focused on 7 of 33 Stage IV melanoma patients who experienced complete and long-lasting remissions following TIL therapy. Using autologous tumor lines (which were prepared for all patients) to stimulate T cells from either the TIL infusion product or from PBMC after “cure”, Sewell found that although the TIL product contained high levels of tumor-specific T cells (usually 10-15%, but up to 44%) as expected, late blood samples still contained detectable, but lower levels (1.6% for the patient whose TILs contained 44% reactive cells). Interestingly, while about 40% of the reactivity observed was due to neoantigens, most (60%) was due to non-mutated shared antigens found in multiple patients, not only with melanoma, but also with other diseases. As an example, the clone Mel8 recognized the Melan A tumor-associated antigen (TAA) found in multiple patients with melanoma. Interestingly, Mel8 also recognized some target in tumors where the Melan A gene was knocked out, or in other diseases that did not express Melan A. To identify these other specificities, paired TCR sequencing allowed quantification and tracking of particular TCRs between TIL and late blood, and thus the identification of interesting orphan TCRs. These were inserted into T cells via CRISPR engineering, and screened using a complete degeneracy arrayed combinatorial peptide library (length-matched for the targeted HLA allele) with expression of MIP-1β as readout, and a novel algorithm (CANTiGEN) to deconvolute the data and identify epitope(s). The approach was validated with multiple T cell clones with known specificity. Application to Mel8 showed that this Melan A-recognizing clone also potentially recognized epitopes from several other non-mutated genes. Two of these, IMP-2 and Bst2, were shown to be endogenously processed epitopes from the full-length genes, and were expressed across a wide range of solid tumor cell lines, explaining how such “multi-pronged TCRs” could recognize a wide variety of cancers. Sewell hypothesized that tumors would be less likely to escape such multi-targeting T cells, and that, based on the number of Mel8 T cells in the infusion product (>1x109) already successfully delivered, such multi-pronged TCRs would be safe. Interestingly, tetramers for the Melan A and IMP-2 epitopes also detected T cells in patients cured of AML or CLL, and were cytotoxic to these cancers in vitro. Mel8 is not the only example, and other multi-pronged TCRs were identified from other patients. This new class of TCRs could form the basis of future TCR-engineered adoptive T cell therapies that may improve sensitivity and potency, be more broadly active, and be less likely to allow immune escape.
Accurate detection of tumor-specific gene fusions with EasyFuse reveals strongly immunogenic personal neo-antigens- Jonas Ibn-Salem - TRON – Translational Oncology at the University Medical Center of Johannes Gutenberg University Mainz Mainz, Germany.
Gene fusions are a source for potentially highly immunogenic personal neoepitopes at the breakpoint, by frameshift or translation of a non-coding region, and can be detected by RNAseq through paired-end or spanning-pair sequencing. However, currently available gene fusion prediction tools lack reliability. When using five different publicly available prediction tools to predict gene fusions in the RNAseq data of 14 triple-negative breast cancers (TNBC), Jonas Ibn-Salem and colleagues observed very little overlap, with the vast majority of gene fusions being predicted by only one tool. While gene fusions predicted by multiple tools showed a higher confirmation rate by qRT-PCR, 61% of gene fusions that were only predicted by one tool could also be validated. To improve the accuracy of gene fusion prediction, Ibn-Salem and team developed a computational pipeline, EasyFuse. EasyFuse includes a read filtering step on paired-end RNAseq data to remove reads that can be mapped to the wild-type transcriptome; fusion prediction by multiple algorithms (FusionCatcher, InFusion, MapSplice2, SOAPfuse, and STAR-Fusion); jointed annotation; stringent re-quantification; and a trained machine learning model. EasyFuse was then applied to predict gene fusion-derived neoantigens in formalin-fixed paraffin-embedded (FFPE) samples from 14 patients with melanoma. Ten of 21 and 1 of 30 predicted fusion neoantigens had spontaneously elicited a CD4+ or CD8+ T cell response, respectively. Across 57 fresh frozen breast cancer samples, a median of 12 fusion neoantigens per sample could be predicted, with 95% of these being non-recurrent. EasyFuse outperformed other published prediction tools and is openly available on Github.
Cancer vaccination strategies
Immune modulatory vaccines in cancer therapy- Mads Hald Andersen - National Center for Cancer Immune Therapy (CCIT-DK), Copenhagen University Hospital, Copenhagen/Herlev, Denmark.
Mads Hald Andersen began by reviewing the decades-long work showing that new vaccination is based not only on targeting cancer cells, but also on effector cells that can recognize and kill suppressive immune regulatory cells that express and present the cognate antigen. Such effectors are called “anti-regulatory T cells” and may significantly modify the suppressive TME in a target-specific manner. Two key cancer cell- and regulatory cell-related molecules include IDO and PD-L1. Work over the years has demonstrated the presence of cytolytic CTLs targeting these antigens in both patients with cancer as well as healthy donors. Moreover, injection of such cells enhanced vaccine-specific immunization and reduced Tregs and IL-10 production. A phase I clinical trial of an IDO vaccine in HLA A02:01-positive, heavily pre-treated patients with non-small cell lung cancer (NSCLC) showed good immunogenicity and a signal of extended survival compared to non-vaccinated, but otherwise similar HLA A02:01-negative patients. Similarly, a phase I trial of vaccination with PD-L1 has led to immune responses to PD-L1. Thus, CTL responses to proteins expressed by various immune suppressive cells may favorably shift the activation/regulatory balance, which might be further enhanced by the stimulatory effects of checkpoint blockade. In the CT26 model, anti-IDO vaccination synergized with anti-PD-1 therapy. Importantly, IDO expression was not from cancer cells, but from myeloid cells in the tumor, leading not to direct killing of tumor cells, but to remodeling of the suppressive TME. A phase I/II trial of biweekly IDO and PD-L1 vaccination combined with anti-PD-1 therapy resulted in an increase in complete and partial responses compared with a strongly matched historical patient cohort. Responses occurred rapidly and no novel safety signals compared to anti-PD-1 alone were observed. Both CD4+ (predominant) and CD8+ T cell responses were observed to both antigens, and TCR sequencing of paired biopsies demonstrated the presence of target-specific cells in the blood and periphery in 4 of 5 patients. Further, target-specific cells could recognize autologous tumor cells or macrophages cultured with tumor cell-conditioned medium to create a TAM phenotype with increased expression of PD-L1 and IDO. Overall, this novel approach opens up multiple new target opportunities, which may be different based on the underlying disease.
Interim analysis of the HIT-HGG Rez Immunvac study -dendritic cell vaccination with partial Treg depletion and double checkpoint blockade in children with relapsed high-grade gliomas- Matthias Eyrich - University Medical Center Würzburg, Würzburg, Germany.
Relapsed glioblastoma in children is a particularly difficult disease to treat, with no approved therapies and a poor prognosis. Key obstacles to immunotherapy options include the presence of regulatory T cells, low immunogenicity due to low mutation load and impaired dendritic cell function, and a hostile TME. Matthias Eyrich described the interim results of the HIT-HGG Rez Immunvac trial that was designed to overcome all these problems. Following surgical removal of the tumor, metronomic, oral cyclophosphamide was administered to deplete regulatory T cells while autologous dendritic cells and a whole-tumor-cell lysate were prepared. Cyclophosphamide was stopped at least 7 days prior to DC/lysate vaccination (4 weekly vaccines doses) followed by dual checkpoint blockade (anti-CTLA-4 + anti-PD-1; 4 doses on a weekly schedule) and then anti-PD-1 alone monthly until 1 year after the initiation of vaccination. The patients were followed for at least 1 year. Nineteen patients were evaluable (8 treated with vaccine only and 11 treated with vaccine plus ICB). The treatment was safe, and although the number of patients was small, the overall survival was encouraging (83% at 6 months) compared to well matched historical controls (62%). Subgroup analysis showed that, as expected, response depended critically on the extent of surgical resection (with partial resection showing the lowest 6-month survival), but was independent of ICB, in line with large trials of ICB in glioblastoma. Stable disease at the conclusion of vaccination was an important positive factor for overall survival. Tregs were effectively depleted during cyclophosphamide therapy. Both CD4+ and CD8+ T cells responsive to in vitro stimulation with tumor lysate were observed, and antigen identification is ongoing.
Dissection of immune responses
Immune-modulation in cancer patients- Jolanda de Vries - Radboud University Medical Center, Nijmegen, Netherlands.
With the aim of understanding the dynamic TME to rationally design new immunotherapy approaches, Jolanda de Vries began by outlining a detailed approach to characterizing the composition (flow cytometry and RNA sequencing) and the gross (tumor, margin, stroma) and detailed spatial location of immune cells in tumors. In particular, de Vries described a high-resolution multiplex IHC approach using spectral imaging and machine learning to count and spatially analyze diverse immune cells. 6 or 7 markers (and DAPI) could be used simultaneously on formalin-fixed paraffin-embedded (FFPE) tissue sections. Analysis of tonsillar tissue validated that the IHC and machine learning analysis accurately detected and quantified the target cell types. Turning to melanoma, de Vries first showed that there was no correlation between the lymphocyte composition of the primary tumor and secondary metastasis from the same patient, but that different metastases were similar, even when separated by many years. Moreover, quantification of cell numbers in primary or metastatic tumors in patients who responded or did not respond to ipilimumab showed no difference. However, there was a clear difference in the tumor mutation burden (TMB) between responders and non-responders, suggesting that TMB was a candidate marker for response to immunotherapy. Lynch Syndrome represents a tumor type with high mutational burden due to mismatch repair defects and predictable frameshift mutations in regions of microsatellite instability. So, a clinical trial was initiated using autologous dendritic cells to deliver frameshift epitopes from TGBβR2 and Casp5, and wild-type CEA, which is frequently overexpressed in colorectal cancer. The vaccine was safe, inducing only mild flu-like symptoms that were higher in patients without ongoing tumor burden, perhaps indicating a more favorable immune state. T cell responses to one or more of the antigens were detected across patients. TGFβR2-targeted T cells that were recovered and expanded from a patient could recognize endogenously expressed mutant, but not the wild-type epitope in a TGFβR2-reconstructed cell line. Casp5-targeted cells could not recognize this line due to low Casp5 expression. Future larger trials with long-term follow-up will be needed to prove clinical efficacy. Automated dendritic cell preparation will be a future key process improvement for this type of vaccine.
Hallmarks of cancer: the immune dichotomy- Douglas Hanahan - Ludwig Institute for Cancer Research Lausanne, Switzerland.
With the extraordinary complexity of cancer at all levels, Hanahan and Weinberg previously rationalized its complexity by defining eight acquired hallmarks in tumors in their landmark paper, Hallmarks of Cancer. In a Keynote address, Douglas Hanahan provided considerations for therapeutic approaches beyond the core concept of these hallmarks. One of the most successful ways the “avoiding immune destruction” hallmark in cancers has been targeted is through immune checkpoint blockade (ICB). However, only a subset of patients respond, and both intrinsic and adaptive resistance mechanisms have been identified. The hallmarks concept can help rationalize immunotherapy, especially with the current wealth of new mechanistic data. A growing armamentarium of “smart drugs” targeting each of the eight hallmarks is available. While targeting one hallmark can have remarkable responses (such as with BRAF inhibitors in metastatic melanoma), tumors generally develop adaptive resistance. The dream scenario would be to target all hallmarks at once, but that would very likely be too toxic. Therefore, combination strategies that are rationally layered and sequenced may achieve improved responses. Hanahan discussed some case studies to illustrate the importance of combining and sequencing therapies. VEGF inhibitors (targeting angiogenesis) and ICB are being successfully combined and used in clinical practice for various cancer types, while ICB combined with BRAF and MEK inhibitors was only found to have a modest benefit that was less effective than combined anti-PD-1 and anti-CTLA-4. However, in the DREAMseq trial, ICB followed by the BRAF/MEK inhibitors provided a better response than the reverse order, and was more effective than double ICB, emphasizing the importance of optimally sequencing therapies. Finally, Hanahan discussed newly defined hallmarks affecting the immune evasion hallmark. First, the microbiome has been found to modulate immunosuppression and antitumor immunity, providing a new dimension to cancer complexity and potential for therapeutic targeting. Second, senescent cells in the tumor microenvironment modulate the immune response through the senescence-associated secretory program (SASP), which can be immune stimulatory or suppressive. Senolytic therapies targeting SASP may provide a therapeutic benefit when combined with immunomodulatory drugs in cases where SASP is immunosuppressive. To close, Hanahan pointed to two further emerging hallmarks, unlocking the phenotypic plasticity of multiple cell types in the tumor microenvironment and epigenetic reprogramming.
Dissecting and modulating reactive anti-tumor immunity in patient-derived tumor fragments- Daniela Thommen - Netherlands Cancer Institute, Amsterdam, The Netherlands.
Daniela Thommen discussed several uses for an interesting ex vivo technology for testing and analyzing the effects of immunotherapies in patient-derived tumor fragments (PDTF). This technology helps to bridge the gap between preclinical and clinical investigations, as it maintains tumor heterogeneity, makes it possible to disrupt certain cell functions, and allows for comparisons of different treatment options on the same tumor. The first example used to highlight the potential of this technology was in dissecting patient responses to PD-1 blockade by seeing if intratumoral T cells could be reinvigorated, and whether this related to response. To test this, Thommen and colleagues analyzed 37 tumor samples, most of which were from primary tumors, while the remainder were from metastases. Fragments were cultured with or without anti-PD-1 antibodies. Analyzing T cell activation markers, chemokines, cytokines, and more, the researchers identified two patterns, correlating with response and non-response to PD-1 blockade. Comparing these patterns to the clinical results observed in 12 patients who received checkpoint blockade therapy, they found that their PDTF platform data aligned perfectly with response and non-response to therapy, suggesting it could be used to predict clinical outcomes. Digging into the question of whether intratumoral T cells can be re-activated by anti-PD-1, the researchers first showed that anti-PD-1 treatment induced cytokine production and cytotoxic markers in responders compared to non-responders. To determine if these were direct effects, they then blocked either LCK (to inhibit T cell activation) or IFNγ (to block downstream effects). In responders to anti-PD-1, blocking IFNγ had minor effects, while blocking LCK prevented the response almost entirely, supporting that the observed changes were a direct effect of anti-PD-1 on T cells. Looking at subsets of T cells that might be associated with response, the researchers found that not all subsets could be reactivated by PD-1. However, while consistent patterns could be identified within patients, the same patterns were not observed between patients. Investigating this heterogeneity using cluster analysis, the researchers identified four patterns of response. The first two were related to strong T cell activation and induction of cytokines, the third was largely a T helper response, and the fourth was a weak response, with low IFNγ and low cytotoxicity despite CD8+ T cell activation. This fourth group showed high levels of OX40 at baseline, possibly reflecting a high Treg compartment. These distinct immunophenotypes may reflect different tumor-reactive T cell pools. Highlighting another use for the PDTF platform, Thommen and team used it to test possible combination therapies. Looking at neoadjuvant checkpoint blockade for treatment of melanoma prior to surgical resection, they found that samples with higher IL-2 signatures were more likely to respond. Testing whether the addition of IL-2 could improve responses to ICB (anti-PD-1 + anti-CTLA-4), they found that samples that responded to ICB also responded to IL-2. More importantly, several samples that did not respond to ICB were converted to responders with the addition of IL-2, suggesting that this therapy could improve responses and increase the fraction of responding patients. This effect was recapitulated in a mouse model of breast cancer, increasing the expansion of tumor-specific T cells and improving responses to neoadjuvant checkpoint blockade. Looking at patient pre-treatment biopsy samples, their system effectively predicted responders to checkpoint blockade, and the addition of IL-2 predicted additional potential responders.
Targeting the tumor immune microenvironment
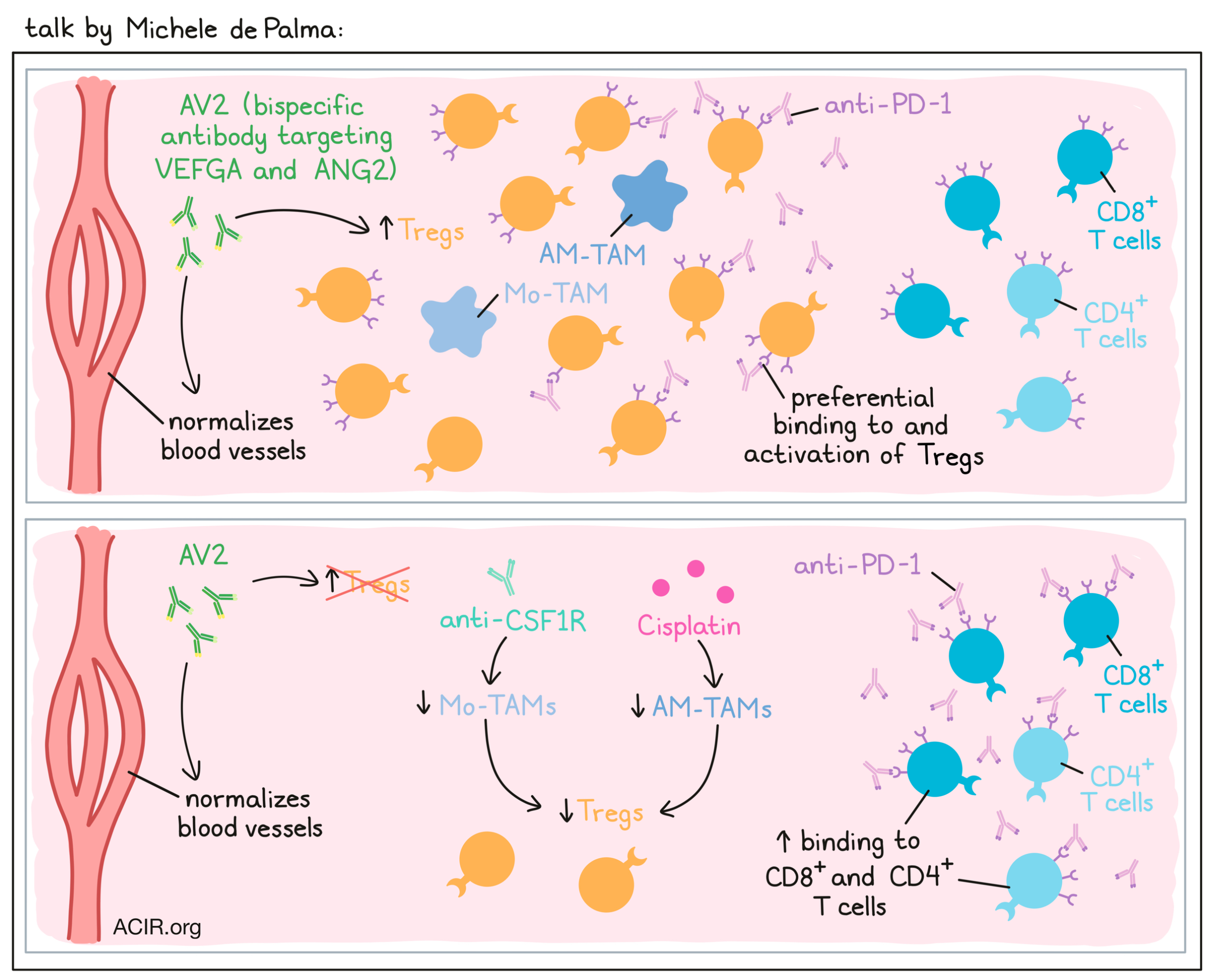
Microenvironmental regulation of tumor responses to immune checkpoint blockade- Michele de Palma - École Polytechnique Fédérale de Lausanne (EFPL), Lausanne.
Michele De Palma discussed recent research on the interplay between tumor blood vessels and T cell infiltration into tumors. Despite the fact that blood vessels in tumors are often leaky and dysfunctional, T cells do not effectively traffic in, likely due to the presence of cytokines that impair extravasation of T cells. For example, molecules such as VEGF result in downregulation of adhesion molecules for T cells on the tumor endothelium. To modulate tumor angiogenesis, De Palma and colleagues utilized a bispecific antibody, AV2, to simultaneously target VEGFA and ANG2 (another angiogenic cytokine), which when blocked together have been shown to normalize blood vessels. AV2 indeed worked to turn “cold” tumors “hot”, increasing CD8+ T cell trafficking and activation, and inhibiting tumors. Given that combined blockade of VEGF is approved for use in combination with PD-1 blockade in the clinic, the researchers tested the addition of anti-PD-1 to AV2. In a KP NSCLC mouse model, AV2 alone induced superior antitumor effects compared to monotherapies directed at either target. Unexpectedly though, the addition of anti-PD-1 to AV2 actually accelerated the progression of tumors. At first, De Palma and colleagues hypothesized that their tumor model might not be sufficiently immunogenic, so they generated a mismatch repair-deficient model with an increased mutation burden and another model that incorporated presentation of OVA, but still the addition of anti-PD-1 failed to synergize with AV2. Next, investigating whether too much genetic variability might be to blame, they focused on a single genetic isolate, but the results remained unchanged, with anti-PD-1 inducing faster tumor progression. Looking at the immune compartment within tumors, the researchers found that AV2 increased Tregs in tumors, that these Tregs robustly expressed PD-1, and that anti-PD-1 antibodies preferentially bound to and activated Tregs over CD8+ T cells or non-regulatory CD4+ T cells. Staining of tissue sections showed that macrophages co-localized with the Tregs, and particularly with PD-1+ Tregs. These macrophages produced IL-10, CCL7, and TGFβ, supporting Treg development. In an effort to inhibit Treg-mediated immunosuppression, De Palma and colleagues tried targeting of macrophages, which can be (mostly) depleted by blocking CSF1R. In addition to lowering macrophages, this also effectively lowered Tregs to baseline levels and improved antitumor immunity in the AV2 + anti-PD-1 treatment group. However, tumors were still progressing on this combination treatment, so to improve responses further, the researchers included the chemotherapy Cisplatin. Interestingly, they found that while CSF1R primarily targeted a group of macrophages called Mo-TAMs, Cisplatin targeted alveolar macrophages (AM-TAMs), which are more proliferative. Together, these two treatments could eliminate the vast majority of macrophages, reducing Tregs even more dramatically. The comprehensive combination of AV2, anti-PD-1, anti-CSF1R, and Cisplatin induced over 70% regressions (in aggregate data). The proposed mechanism is that elimination of macrophages supports the reduction of Tregs, which reduces competition for anti-PD-1 antibodies and allows for effective CD8+ T cell activation. This model fits with observations that high levels of macrophages are often accompanied by high levels of Tregs in human NSCLC, and observations that some patients with lung cancer experience hyperprogression on PD-1 therapies.
A novel role of IL-10 in maintaining anti-tumor immunity- Martina Seiffert - German Cancer Research Center (DKFZ), Heidelberg, Germany.
Martina Seiffert used the Eμ-TCL1 model of B-cell lymphoma to track down the mechanism of immune evasion, and uncovered a novel role for IL-10. In this model, high IDO expression and accumulation of the inhibitory metabolite kynurenine suggested that IDO1 inhibition might be effective against the cancer. However, epacadostat, an IDO inhibitor, had no effect in the Eμ-TCL1 model, mirroring the lack of effect observed in the large phase 3 trial of this drug in combination with anti-PD-1 therapy that led to discontinuation of development of the drug. Searching for other molecules that might be involved in the activation of AHR, a key receptor-linked transcription factor involved in inhibiting CD8+ T cell activity, an extensive analysis of TCGA expression data across multiple tumor types, coupled with natural language processing review of literature revealed that Il4i1 was more closely associated with AHR activity than IDO1 or other suspected additional targets (IDO2, TDO2). Il4i1 is a secreted oxidase that acts on multiple amino acids to produce various metabolites that can be agonists for AHR. It is produced by monocytes and is found in high levels in the serum of Eμ-TCL1 compared to wild-type mice. Knockout of Il4i1 slowed growth of Eμ-TCL1 cells and generated less exhausted CD8+ T cells with an intermediate (versus high) PD-1 expression level, pointing to therapeutic potential of Il4i1 inhibition. Digging deeper, knockout of AHR in the model was not as effective as blockade of IL4i1, suggesting that other mechanisms were also driving exhaustion. Single-cell RNA sequencing of CD8+ T cells from Eμ-TCL1 mice revealed the similarities of the PD-1high cells to classical exhausted cells, and a distinction from PD-1-intermediate cells, which appeared more active. The RNA data also revealed differential cytokine and cytokine receptor expression, and, in particular, differential upregulation of IL-10 and downregulation of IL-10R in the PD-1-high versus -intermediate cells, respectively. Given the described role of IL-10 as an immune-dampening cytokine, Seiffert blocked IL-10R and, surprisingly, found that tumors progressed more rapidly and that CD8+ T cells became more exhausted. This observation was confirmed with IL-10R and STAT3 knockouts, which mechanistically affected the NFAT/AP-1 transcription factor balance, resulting in differential chromatin accessibilities of critical activation and exhaustion genes. Thus, IL-10 signaling is important for a favorable balance of active PD-1-intermediate cells. In patients with CLL, high IL-10 expression was correlated with better survival. Moreover, PD-1-high cells accumulated in lymph nodes of patients with CLL and showed a higher expression of exhaustion and Treg markers in malignant lymph nodes, pointing to the potential to positively modulate IL-10 as a novel therapeutic mechanism.
Tumor-derived GDF-15 prevents therapy success of checkpoint inhibitors by blocking T-lymphocyte recruitment- Jӧrg Wischhusen - CatalYm GmbH, Planegg-Martinsried, Germany.
Beginning with the analogy that fetal and tumor growth both require a host-intrinsic tolerance mechanism to survive, Jӧrg Wischhusen then showed that the level of GDF-15 was lower in the serum of female patients who had miscarriages than in those who had viable deliveries – a result confirmed in mouse models. GDF-15 is a member of the TGFβ super-family that binds to the extracellular matrix and is released into serum, and which has high expression levels in placenta, prostate, and many solid tumors. Earlier data showed that high GDF-15 correlates with poor survival in multiple tumor types. Mechanistically, as T cell extravasation to tissues requires interactions with endothelial cells, Wischhusen conducted an unbiased screen to look for the effects of GDF-15 on activated endothelial cell adhesion and found that CD4+, CD8+ and NK cell binding were all reduced in the presence of GDF-15. The interaction pathway for T cells was through LFA-1 on T cells and ICAM1 on endothelial cells, including lymphatic endothelial cells, and was associated with lower T cell infiltration in melanoma and HPV+ oropharyngeal squamous cell carcinoma (OPSCC). A MC38 mouse model showed that overexpression of GDF-15 in tumor cells accelerated tumor growth and reduced T cell infiltration, and that antibody blockade improved the effectiveness of immunotherapy (agonist CD40 + pIC or anti-PD-1). Similar results were seen in an orthotopic pancreatic tumor model and a PDX model. Interestingly, in patients with melanoma who were treated with anti-CTLA-4, although initial responses did not differ between low and high GDF-15 expression, durable responses were associated with low expression, indicating GDF-15 levels are a predictor of durable response. Similarly, low GDF-15 levels were predictive of overall survival following anti-PD-1 therapy, even for patients with high LDH (indicative of large tumor burden). Wischhusen summarized by indicating that neutralizing GDF-15 in patients may be a viable and useful therapeutic approach, and that phase 2 trials are now underway with visugromab.
Synergistic effects of intratumoral mRNA transfer of single chain interleukin 12 and a decoy-resistant variant of interleukin 18- Assunta Cirella - Universidad de Navarra Pamplona, Spain.
Although multiple approaches have been designed to overcome the toxicity of systemic IL-12 therapy due to IFNγ upregulation, more options are needed. IL-12 upregulates the expression of the IL-18 receptor complex, and IL-12 and IL-18 synergize in IFNγ induction, but with increased toxicity when delivered systemically. In an attempt to capitalize on this synergy, Assunta Cirella turned to an approach involving intratumoral expression of these cytokines from transient mRNA. However, the tumor microenvironment is enriched in IL-18 binding protein, a decoy receptor that binds to and abrogates IL-18 activity. To overcome this blockade of IL-18 activity, Cirella used a modified IL-18 molecule, DR-18, that is “decoy-resistant” (i.e., does not bind to the IL-18 binding protein). When mRNA was delivered via TransIT®, in vitro expression of both cytokines peaked at 48-72 hours. Intravenous injection of TransIT® -formulated IL-12 + DR-18 RNA combination, but not of IL-12 + IL-18 RNA combination, revealed liver toxicity, indicating stronger IL-18 activity with DR-18. Intratumoral injection of unformulated luciferase mRNA demonstrated expression in B16F10-OVA tumors and multiple stromal cells; intratumoral injection of IL-12 + DR-18 led to comparable tumor control to IL-12 + IL-18, but tumor growth in the contralateral tumor was more inhibited by IL-12 + DR-18. Control on both sides was dependent on CD8+ T cells, and the contralateral tumor demonstrated multiple signs of CD8+ T cell activation. IL-12 + DR-18 also synergized with anti-PD-1 therapy, inducing responses in the contralateral non-injected tumor. These results suggest a potential pathway to maximize the efficacy of IL-12 by combination with an IL-18 variant while limiting toxicity via intratumoral expression of transient mRNA.
By Lauren Hitchings, Maartje Wouters, Ed Fritsch, and Ute Burkhard.



