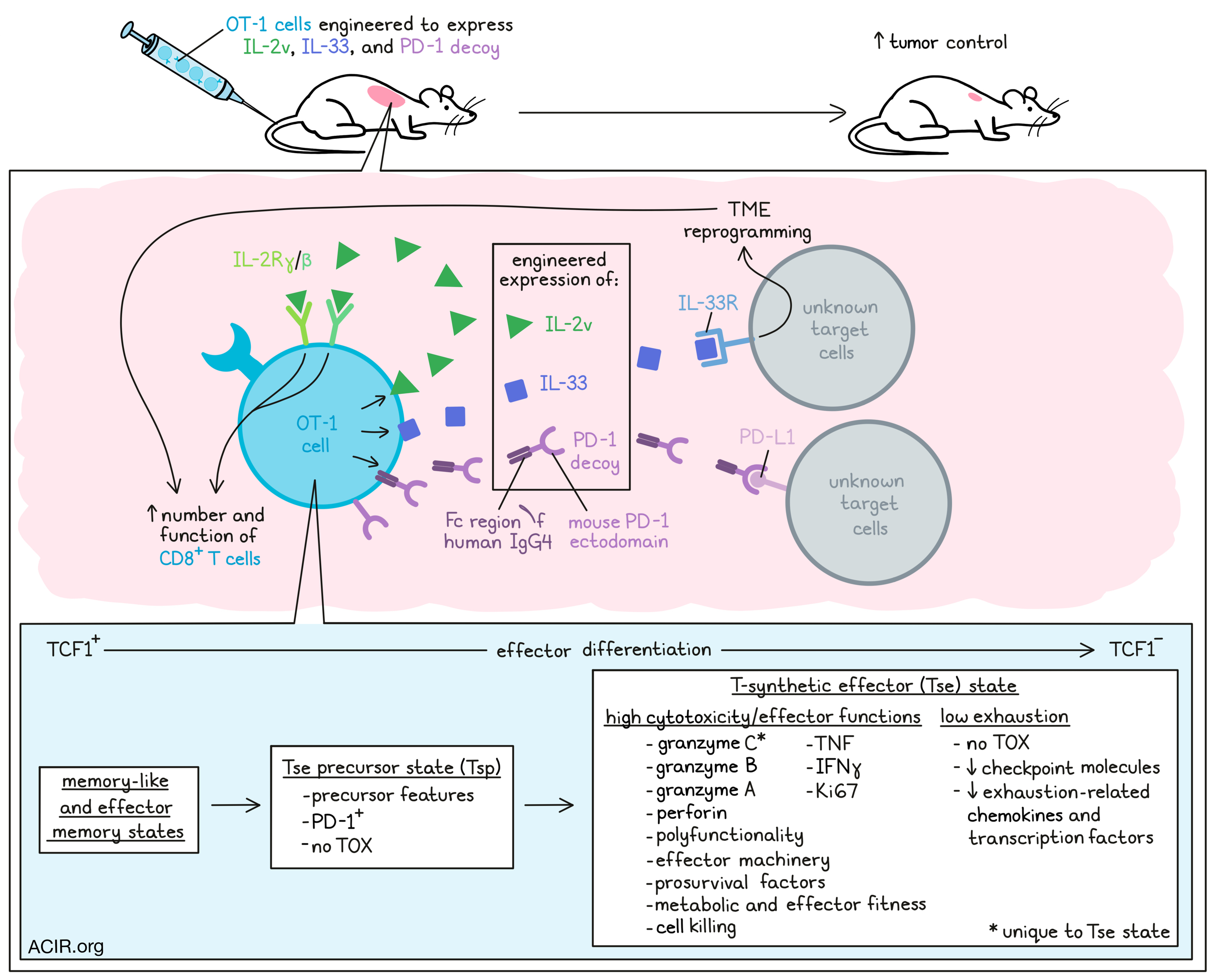
T cell exhaustion in response to chronic activation of effector CD8+ T cells limits the efficacy of particular adoptive cell transfer (ACT) therapies. Corria-Osora et al. hypothesized that T cells could be rewired by engineering them to express orthogonal cytokines to acquire and maintain a functional state upon chronic stimulation. The researchers assessed various engineered T cells to research this hypothesis in murine models, from which the data were recently published in Nature Immunology.
The researchers engineered OT-1 T cells to express an IL-2 variant (IL-2v) that does not engage with the IL-2 receptor α-chain, as well as IL-33, which serves as a pro-inflammatory alarmin. Furthermore, a secreted PD-1 decoy (PD1d – a fusion molecule of the ectodomain of mouse PD-1 with the Fc region of human IgG4) was also included. Cells engineered with individual molecules, PD1d/IL-2v, PD1d/IL-33, or PD1d/IL-2v/IL-33 were shown to secrete the relevant transgenes.
Mice with advanced non-immunogenic B16-OVA tumors were treated with ACT with various OT-1 variants without preconditioning lymphodepletion. Untransduced, PD1d, IL-2v, or PD1d/IL-2v-transduced OT-1 cells had minimal impact on tumor growth. Transduction with IL-33 alone or PD1d/IL-33 had limited effects on tumor growth. However, PD1d/IL-2v/IL-33 OT-1 cells induced tumor regression and resulted in an objective response rate (ORR) of 85.7%, compared to 0-9% for all other cell variants. When ACT was administered earlier, therapy cured the mice.
There was a significant increase in CD8+ T cells in the tumor in response to PD1d/IL-2v/IL-33 OT-1 cell ACT, as compared to ACT with the other OT-1 variants. Phenotypically, when IL-2v was included in the cells, it resulted in expansion of TCF1+ OT-1 cells. High numbers of TCF1- effector CD8+ TILs, necessary for tumor regression, were only detected after ACT with PD1d/IL-2v/IL-33 OT-1 cells, suggesting that IL-33 was necessary for TCF1+ precursor cells to transition to effector cells.
Total CD8+ TILs were isolated from B16-OVA tumors treated with each ACT subset. Single-cell RNAseq revealed five distinctive clusters. PD1d/IL-2v/IL-33 cells clustered separately (C5) from canonical T cell clusters, and were characterized by low levels of exhaustion markers and high levels of cytotoxicity genes, including Gzmb and, uniquely, Gzmc. Projecting these clusters on a reference atlas suggested cells in cluster C5 were projected to canonical Tex. To further assess the C5 TILs, they were subjected to an independent higher-dimension analysis. This separated the C5 TILs from the reference Tex TILs, as the C5 cells had a unique cytotoxic effector-like program, with granzymes, perforin, and effector machinery genes and prosurvival factors. These data suggested these cells were different from the TOX+ exhausted phenotype, as these cells did not express TOX, and had downregulated expression of checkpoint molecules and various exhaustion-associated chemokines and transcription factors.
Based on these data, the researchers suggest that the C5 cells represented a novel T-synthetic effector (Tse) state. When OT-1 and endogenous CD44+CD8+ TILs were analyzed separately after PD1d/IL-2v/IL-33 ACT, two new clusters were detected (C6 and C7). The endogenous TILs mostly clustered in the C4 and C2 clusters during tumor regression after PD1d/IL-2v/IL-33 ACT, and the C5 Tse cells were specific for OT-1 TILs. This C5 state was gradually acquired during tumor regression. Most OT-1 cells on day 5 after ACT were distributed in the C2, C6, and C7 clusters, with minimal in C5, while on day 8, most of these cells clustered in C5 and C6, and by day 12, almost all clustered in C5.
After building a new TIL reference map using the obtained data, the researchers assessed whether this novel effector state was detectable in other T cell datasets. They did not detect this profile in various tumor cohorts, datasets obtained after ICB, or CD19 CAR-T datasets. Tse TIL were also different from previously described CD8+ “better effector” (Tbe) TILs, confirming that the C5 Tse might be a novel and synthetic effector state.
To further characterize the C5 Tse state, TILs were phenotyped by gating on TOX and Gzmc. In untreated mice, CD8+ TILs had a TOX+Gzmclo-neg exhausted phenotype. During tumor regression after PD1d/IL-2v/IL-33 ACT, OT-1 cells expressed PD-1 and were enriched in TOX-Gzmc+ cells, with most Gzmc+ cells being TCF1-. These cells were polyfunctional effector cells, expressing Gzmb, Gzma, perforin, TNF, IFNγ, and Ki67. Furthermore, they were enriched for genes associated with energy production, effector molecule synthesis, and cell killing, suggestive of metabolic and effector fitness.
To assess the role PD-1 functionality plays in the Tse TILs, the researchers added anti-PD-L1 treatment to PD1d/IL-2v/IL-33 ACT, and found that it did not improve tumor control in response to ACT. Furthermore, removing the PD-1 ectodomain from the PD1d did not impact tumor control of IL-2v/IL-33-transduced OT-1 cells, nor did it impair accumulation of PD-1+Gzmc+TCF1- OT-1 TIL. These data suggest that the Tse TIL were not dependent on PD-1-mediated inhibition.
Gzmc+ cells were not detected in the tumor-draining lymph nodes (TDLN) or spleens after ACT, suggesting the Tse state was specific to tumors. As Tse TIL did not express the IL-33 receptor, the contribution of IL-33 was likely through indirect reprogramming of the tumor microenvironment.
The researchers found that the C6 cluster cells were characterized by precursor features, expression of Pdcd1 and stemness-related markers, and overexpression of Gzmc and other effector molecules, while cells were negative for TOX. Therefore, these cells might represent a Tse precursor state (Tsp). Trajectory and pseudotime analyses suggested that the OT-1 cells transitioned from memory-like and effector memory states to Tsp and then Tse.
To assess the role IL-2v played in the development of these various T cell states, OT-1 TILs obtained 12 days after IL-2v OT-1 ACT were analyzed. These cells mostly clustered in the C7 memory-like cluster, which was characterized by high expression of T cell activation and stemness-related markers, low expression of checkpoint molecules, and a lack of TOX and Gzmc expression. Therefore, IL-2v may play a role in preparing cells for the superior effector function of Tse and Tsp cells. Transcription factor analysis revealed that IL-2v was involved in the regulation of two sets of transcription factors – one that was active in the C7 TILs in the absence of IL-33, associated with sustaining cell expansion and persistence, stemness, and memory/readiness, and a second that was active across all synthetic states, involved in metabolic support.
Together, these data suggest that by engineering T cells to express IL-2v and IL-33, with or without the PD-1 decoy PD1d, a new functional subset of T cells can be obtained that is activated in the tumor microenvironment and effectively targets murine tumors, but without antigen-driven exhaustion. If these data can be reproduced in human TILs, this may be a strategy to overcome some of the current limitations of ACT therapy in patients.
Written by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, first author Jesus Corria-Osorio answered our questions.

What was the most surprising finding of this study for you?
That IL-2v/IL-33-engineered T cells evade TOX+ canonical exhaustion and acquire an improved functional effector state in tumors, achieving high levels of engraftment and driving tumor regression without the need for prior lymphodepletion or exogenous cytokine support post-adoptive T cell therapy (ACT). This novel PD-1+TOX- T cell synthetic effector (TSE) state was not functionally restrained by the PD-1/PD-L1 axis, and has seemingly never been observed naturally in mice or humans to date.
What is the outlook?
This discovery offers new insights about the plasticity of CD8+ TIL states and unlocks the opportunity to rationally develop novel “synthetic” T cell states through genetic engineering. These cells were not only very potent, but expanded in a cell-autonomous manner, engrafted the host, and expanded in tumors without the need for lymphodepletion or IL-2 support. If we reproduced this TSE state with engineered T cells in the clinic, we would have a more effective ACT approach for solid tumors, and it could lead to substantial improvement of toxicities and clinical care costs associated with ACT, particularly for ACT-based TILs, in which profound lymphodepletion and adjuvant IL-2 administration are responsible for most toxicities.
What was the coolest thing you’ve learned (about) recently outside of work?
I recently discovered a very cool sport called Paddle that is very similar to Tennis. I love it. It makes me super happy these days.



