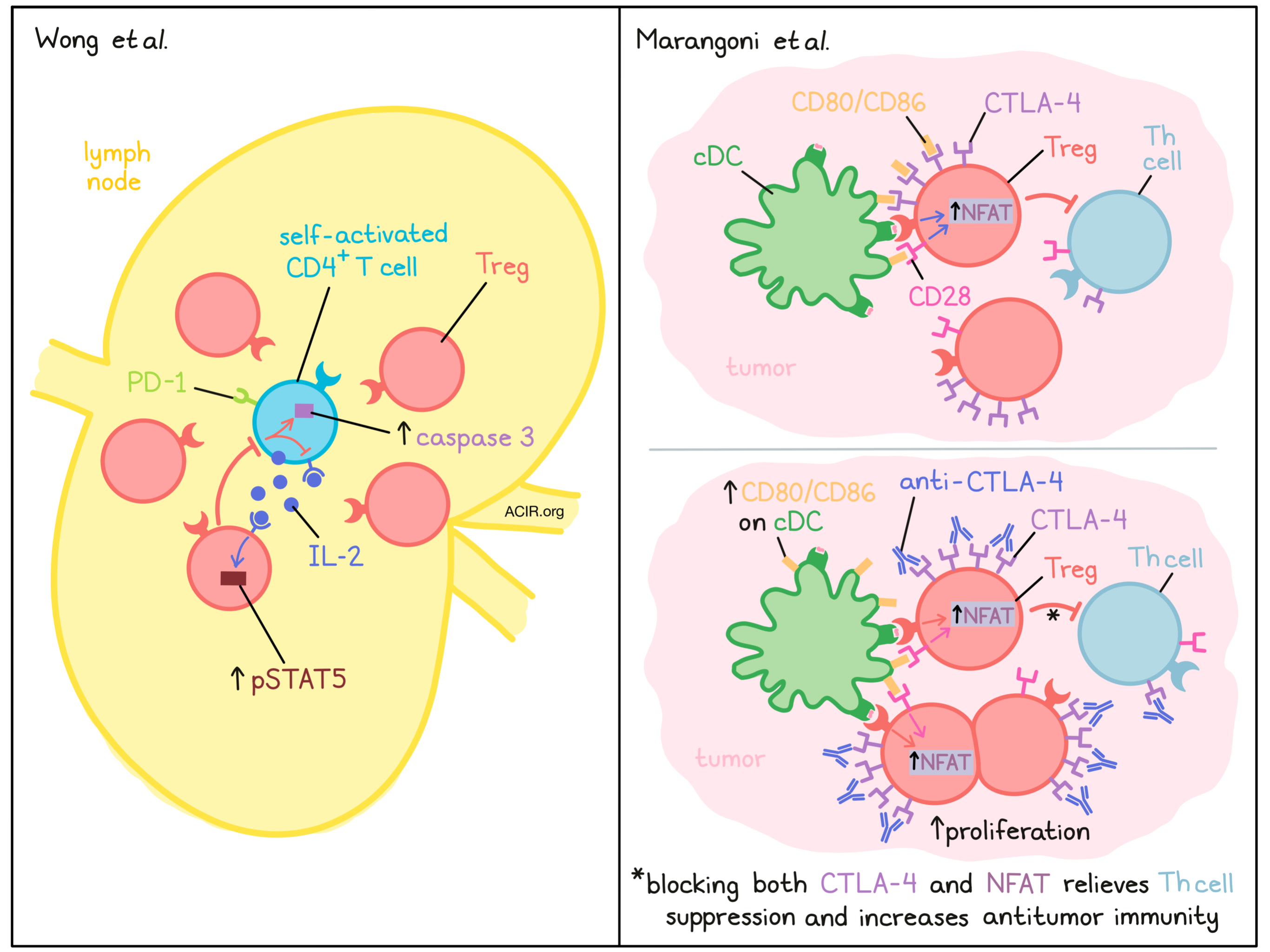
Although Tregs maintain homeostasis and fend off autoimmunity, they can also dampen the antitumor immune response. Despite these major roles, key facets of Treg biology have remained unknown. For instance, how exactly do Tregs constrain self-activated T cell activity in secondary lymphoid organs? And what regulates Treg abundance and function in the TME? In two papers recently published in Cell, Wong et al. and Marangoni et al. addressed these respective questions, providing novel insights into Treg function.
Wong et al. focused on uncovering the mechanisms underlying Treg control of self-activated T cells. Staining lymph nodes (LNs) of healthy C57 mice, they observed heterogeneous, but cell-focused expression of PD-1 among CD4+ T cells, implying TCR signaling (i.e. self-antigen recognition). Interestingly, these PD-1+CD4+ T cells localized with Treg-dense “micro-domains,” enriched for proliferating, effector-type Tregs expressing pSTAT5, indicating IL-2 receptor signaling. The researchers theorized that the PD-1+CD4+ T cells may be the source of this IL-2, and indeed, antibody blockade of IL-2 reduced Treg density around PD-1+CD4+ T cells and Treg activation in micro-domains, more so than other locations in the LN. Furthermore, disrupting Treg micro-domains by Treg protein synthesis inhibition in Foxp3-DTR mice increased pSTAT5+CD4+ T cells, which were more enriched in PD-1 expression than pSTAT5- cells. These results suggested that IL-2 produced by self-activated T cells leads to the increased Treg density in the surrounding region.
The researchers next questioned the fate of the self-activated T cells interacting with Treg micro-domains. The team transferred TxA23 cells, transgenic CD4+ T cells specific for a gastric self-antigen, along with wild-type cells, into healthy BALB/c mice. In gastric LNs, TxA23, but not wild-type cells upregulated PD-1 and proliferated. However, this process was only transient, and their numbers soon dropped. Additional evidence for this “pruning” effect was found in the PD-1+CD4+ T cells in C57 mice (where the target antigens are unknown) which expressed high levels of the apoptotic marker caspase 3. Furthermore, boosting self-activated T cell IL-2 signaling through an IL-2Rβ/γ-engaging immunocomplex, which stimulates conventional CD4+ T cells, but not Tregs, reduced caspase 3 expression in PD-1+ (but not PD-1-) CD4+ T cells in C57 mice, and increased TxA23 cell number in the BALB/c transfer model, supporting their survival. Overall, these findings suggest that Tregs curb the IL-2 signaling needed for self-activated T cell proliferation.
Because many parameters could influence the fate of self-activated T cells (pruning vs. escape), Wong et al. developed a computational model to probe perturbations in these variables, using self-activated T cell pSTAT5 expression as a readout. The model predicted that small changes in Treg micro-domain size, CTLA-4 functionality, or TCR-ligand affinity, for example, could significantly alter the pSTAT5 expression of self-activated T cells. Interestingly, the model predicted non-linear relationships between certain variables, and in vivo results matched these predictions. In one example, the researchers generated bone marrow chimeras with varying degrees of Treg ablation. While a 15% decrease in Treg density did not alter pSTAT5 expression in T cells, a 40% Treg reduction significantly increased pSTAT5+CD4+ T cells and decreased caspase 3 expression, as predicted. Increasing self-activated TCR–ligand affinity, another variable, increased pSTAT5 expression both in silico and in vivo. These findings contextualized the variables involved in Treg oversight, and provided a framework to consider which interactions may result in tolerance or autoimmunity.
While Wong and coworkers considered Treg behavior in LNs, Marangoni et al. investigated Treg populations within the TME. Their team used intravital microscopy to image Tregs or CD4+Foxp3- T helper (Th) cells transduced with fluorescent markers tagging nuclei and NFAT (to indicate TCR activation). These cells were transferred into CD11c reporter mice (to mark APCs), and were observed within the MC38 TME. Compared to Th cells, Tregs had greater NFAT signaling, which began in proximity with CD11c+ APCs. Tregs also migrated more slowly around APCs, overall suggesting presentation of Treg antigens within the TME. Seeking to uncover which APC subset was responsible, the researchers ablated cDCs (zbtb46-DTR mice) and found reduced Treg NFAT activation and increased migratory speed. Importantly, cDC loss reduced Treg cell numbers in the TME – a response mimicked by blocking NFAT activation in Tregs, suggesting that TCR engagement with cDCs maintains Tregs in the TME.
To eliminate differences between Foxp3+ and Foxp3- cells that could arise solely due to differences in TCR avidity, due to recognition of self-antigens by thymic-derived Tregs versus foreign antigens by Foxp3- CD4+ Th cells, the researchers compared behavior of Treg and Th cells derived from influenza hemagglutinin (HA) TCR-transgenic mice within the CT26 TME. As expected, NFAT activation was comparable, but NFAT-signaling Tregs had less stable contacts with APCs than NFAT-signaling Th cells, indicating that this effect was cell-type- and not TCR-intrinsic. Interestingly, however, Th cells that were co-transferred with Tregs reduced their APC contact stability relative to Th cells transferred alone, suggesting that Tregs can broadly destabilize Th-APC contacts.
The team next considered how Treg behavior changes in the context of anti-CTLA-4 immunotherapy. Tregs express high levels of CTLA-4, which has been shown to be capable of removal of the co-stimulatory molecules CD80 and CD86 from the surface of APCs, potentially abrogating their stimulatory capability. Administering a blocking, but non-Treg-depleting anti-CTLA-4 mAb, the researchers found increased CD80/CD86 expression on TME cDC1s. A similar increase was observed upon Treg ablation (Foxp3-DTR model), indicating that Tregs may reduce APC costimulation via CTLA-4. Interestingly, CTLA-4 blockade also increased Treg–APC engagement and Treg abundance and proliferation within the TME, implying that high CTLA-4 expression on Tregs reduced DC costimulation via lowering CD80/86 on APCs, and that Tregs can limit their own proliferation as a negative feedback loop.
The authors further probed the mechanisms involved, first questioning whether CTLA-4 acted in cis (in a cell-intrinsic manner) or trans (by influencing other cells) to resist Treg expansion. Selective genetic knockout of CTLA-4 in a small sub-population of Tregs did not elicit their proliferation, suggesting that CTLA-4 from the larger CTLA-4-containing population acted in trans. Mechanistically, Treg proliferation following anti-CTLA-4 treatment was not due to IL-2, as anti-CTLA-4 induced proliferation with or without IL-2Rα/β blockade. The authors next theorized that given the ability of CTLA-4 to impact CD80/86 levels on APCs, direct costimulation, through CD28, could drive Treg proliferation. Strikingly, in mice where CD28 was selectively ablated, CD28lo Tregs did not proliferate in response to anti-CTLA-4, unlike CD28hi cells. Thus, APC costimulation could maintain TME Treg populations through CD28, and Tregs, which can modulate co-stimulatory CD80/86 on APCs, can self-regulate.
Finally, the researchers considered whether Treg expansion after anti-CTLA-4 therapy restrains antitumor immunity, even when CTLA-4 binding to CD80/86 is blocked, but Tregs are not depleted. Although MC38 tumors still progressed after anti-CTLA-4 therapy, which did not deplete Tregs, combining anti-CTLA-4 with Treg NFAT inactivation improved tumor rejection, confirming that Tregs still suppress antitumor immunity even in the context of CTLA-4 blockade.
Altogether, these papers provided intriguing insights into Treg biology. Wong et al. introduced a feedback loop in which self-activated T cells produce IL-2, increasing nearby Treg density to cull their proliferation, and proposed that multiple factors act in concert to determine whether this control is lost. Marangoni et al. found that TME Tregs regulate their abundance by modulating the CD28 costimulation they receive from CD80/CD86 on cDCs, and this barrier is lifted by CTLA-4 blockade. Broadly, uncovering the immunological mechanisms underlying Treg behavior in lymphoid organs and tumors can support the development of novel cancer immunotherapies and inform combination strategies.
Write-up by Alex Najibi, image by Lauren Hitching
Meet the researcher
This week, the first authors on both papers, Harikesh Wong (not pictured) and Francesco Marangoni, answered our questions.

What prompted you to tackle this research question?
HW: It was long appreciated that regulatory T cells (Tregs) prevented host autoimmunity mediated by conventional T cells. However, Ron (Germain) and I felt that it was unclear how Tregs operated: how did they selectively limit autoimmune T cell responses while simultaneously enabling host-protective responses against pathogens? Although Tregs had been studied extensively at the molecular level, a framework explaining their operations in complex tissue environments – where cells communicate in a highly coordinated manner – was lacking. We felt that understanding communication dynamics between Tregs and activated T cells would provide critical insights into how the earliest stages of autoimmunity were initiated and controlled.
FM: I was amazed by the benefits that checkpoint blockade immunotherapy gave to many melanoma patients. During a seminar, the Nobel Prize winner Jim Allison told the story of a young lady diagnosed with metastatic melanoma in 2005 – at that point in time, it was almost a death sentence. She was treated by Prof. Allison with CTLA-4-blocking agents, getting complete remission. As of today, she is a healthy woman and a mother. Unfortunately, however, checkpoint blockade immunotherapy does not work this well in all patients, so it is crucial to understand the mechanisms by which it functions in detail, to remove the hurdles that prevent its full efficacy and benefit as many patients as possible. Immunotherapy is not selective, meaning that when it is administered to patients, it does not target solely the immune cells that would cause tumor rejection, but the whole immune system. The scientific question that sparked my curiosity was “what about if checkpoint blockade also triggers immune mechanisms that do not go in the best interest of the patients?".
What was the most surprising finding of this study for you?
HW: We were most surprised to find that T cells activated by self-antigens underwent proliferation, even in healthy hosts. We knew that some of these cells could cause autoimmune tissue damage, so seeing them proliferate in the presence of perfectly functional Tregs came as an initial shock. However, we eventually found that the observed proliferation was quite short-lived, and that self-antigen-activated T cells died rapidly, a process we referred to as “pruning”. Pruning occurred because Tregs accumulated around each activated T cell, preventing the latter from receiving signals required for a productive response. Notably, these local constraints were initiated in response to T cell activation, not prior, revealing a negative feedback circuit. We also found that relatively small changes in Treg function enabled a significant fraction of self-antigen-activated T cells to escape pruning and become threats to the host. This observation was equally surprising to us because it demonstrated how easily the circuit could be imbalanced. We believe this finding is highly relevant for understanding how autoimmune diseases emerge naturally and why immunotherapies used to treat cancer often produce autoimmune side effects.
FM: We started our study characterizing the activation of tumor-associated T regulatory cells, a key immunosuppressive cell type, using intravital microscopy. I was so surprised to see that Treg cells are activated by dendritic cells in tumors even more frequently than protective effector T cells. Activated Treg cells pruned costimulatory B7 molecules on dendritic cells via CTLA-4, in turn decreasing the amount of co-stimulation they themselves received, decreasing their numbers. Administration of CTLA-4 blockers increased the availability of B7 molecules on dendritic cells and expanded tumor-associated Treg cells even more than effector T cells. The most surprising finding was that these expanded Treg cells, despite the fact that they were deprived of a key mechanism of immunosuppression (CTLA-4), still inhibited tumor rejection through alternative mechanisms. Thus, we found that CTLA-4 blockade immunotherapy activates its own brakes. In the future, it will be crucial to remove these brakes in order to benefit as many patients possible.
What was the coolest thing you’ve learned (about) recently outside of work?
HW: My life has changed a lot recently, because I became a dad. My son is providing me with new excitement on a daily basis and a new perspective on life.
FM: It was truly awesome to be able to taste life again after this difficult pandemic year. When my family and I started to attend kids’ birthday parties again, or doing little trips on Sundays, or getting an ice cream all together, or having friends to visit us, it was like catching a breath of fresh air after so much time. It definitely reminds me of the infinite importance of small things, which so many times before the pandemic, I overlooked.




