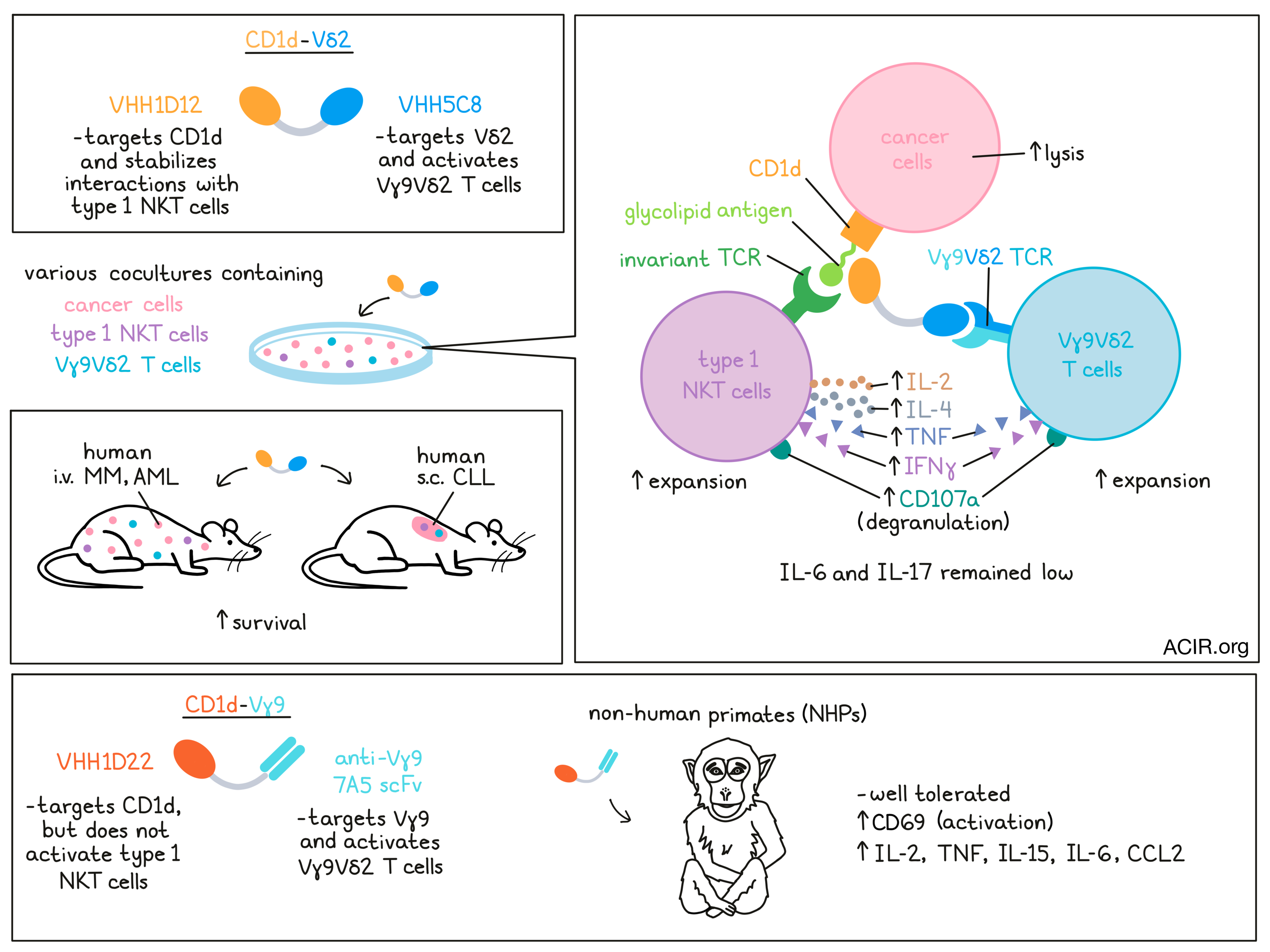
Bispecific T cell engagers (bsTCE) are powerful immunotherapeutic tools, but they come with issues of toxicity and Treg activation. In an effort to overcome some of these challenges, Lameris et al. developed a CD1d-Vδ2 bsTCE to activate both type 1 NKT cells and Vγ9Vδ2-T cells against cancers expressing CD1d – a non-polymorphic MHC-I-like molecule that presents self and foreign glycolipid antigens – including multiple myeloma (MM), myelomonocytic acute myeloid leukemia (AML), and chronic lymphocytic leukemia (CLL). Their results were published in Cell Reports Medicine.
To generate this bsTCE for use in mice, the researchers combined two single-domain antibodies: VHH1D12, which recognizes the complex between glycolipid-bound CD1d and the invariant TCR found on NKT cells; and VHH5C8, which recognizes the Vδ2 portion of the TCR on Vγ9Vδ2-T cells. The resulting bispecific molecule, CD1d-Vδ2, shows trispecific properties, as it simultaneously engages Vγ9Vδ2-T cells with cancer cells expressing CD1d, and stabilizes the interactions between type 1 NKT cells and lipid antigen-bound CD1d, enabling strong responses from both Vγ9Vδ2-T cells and type 1 NKT cells against CD1d+ cancers.
In cocultures of type 1 NKT cells and/or Vγ9Vδ2-T cells with various CD1d+ tumor cell lines, CD1d-Vδ2 induced upregulation of the degranulation marker CD107a on both type 1 NKT cells and Vγ9Vδ2-T cells. Type 1 NKT cells secreted Th1- and Th2-associated cytokines, including IL-2, IL-4, TNF, and IFNγ, while Vγ9Vδ2-T cells mainly secreted TNF and IFNγ. Tumor cell lysis was observed across all CD1d+ (but not CD1d-) tumor cell lines, even at very low effector:target cell ratios. When the type 1 NKT cells and/or Vγ9Vδ2-T cells were swapped out for PBMCs, CD1d-Vδ2 promoted strong type 1 NKT cell and Vγ9Vδ2-T cell expansion, and mediated control over tumor cell growth. Limited cytotoxic activity was observed against healthy B cells or monocytes, which also express CD1d.
Next, the researchers developed a humanized version of CD1d-Vδ2 by generating and analyzing humanized sequence variants of CD1d VHH1D12 and Vδ2 VHH5C8. While humanized Vδ2 VHH5C8 showed similar binding to the Vγ9Vδ2 TCR, all of the humanized CD1d VHH1D12 variants had reduced CD1d binding and reduced effects on iNKT cells. The most favorable variants of each VHH were selected and linked.
Like their murine counterparts, the humanized bsTCEs effectively promoted expansion of type 1 NKT cells and Vγ9Vδ2-T cells, which could be further boosted with the addition of IL-2. They also induced antitumor activity in bone marrow mononuclear cell (BMMC) samples from patients with MM or monocytic AML, and in PBMCs of patients with CLL. While degranulation of patient Vγ9Vδ2-T cells was clearly evident, degranulation of autologous type 1 NKT cells could not be confirmed due to their very low frequency in the tumor samples. When cancer samples were mixed with healthy donor-derived type 1 NKT cells, Vγ9Vδ2-T cells, or a 1:1 mixture of the two, CD1d-Vδ2 hu-bsTCE induced robust degranulation and cytokine production from both cell types, resulting in cancer cell lysis. Notably, IL-6, a known driver of cytokine release syndrome (CRS) remained low, as did IL-17. Interestingly, CD1d-Vδ2 only triggered minimal type 1 NKT cell activation and cytolytic activity against CLL cells. This was found to be due to a failure in lipid antigen loading onto CD1d in CLL cells.
To investigate the effects of CD1d-Vδ2 in vivo, Lameris et al. established mouse models of systemic human AML and MM in immunodeficient NSG mice. In the AML model, which grew aggressively, i.v. administration of mixed (1:1) human type 1 NKT and Vγ9Vδ2-T cells at day 0 and 7 had a modest effect on survival. However, the addition of CD1d-Vδ2, administered twice weekly, significantly prolonged survival. Similarly, in the less aggressive MM model, human type 1 NKT cells and Vγ9Vδ2-T cells administered on days 7, 14, and 21 extended survival, but all mice ultimately succumbed to their tumors. With the addition of CD1d-Vδ2 bsTCEs though, 7 out of 8 mice survived.
Next, Lemaris et al. evaluated the in vivo antitumor efficacy of the humanized CD1d-Vδ2 bsTCE in a s.c. T-ALL model in NSG mice. Tumors were established using either CCRF-CEM cells (T-ALL) or CCRF-CEM cells admixed with whole PBMCs. While the PBMCs themselves slightly reduced tumor growth, the addition of CD1d-Vδ2 strongly inhibited tumor growth in a dose-dependent manner, with tumor growth only occurring upon the discontinuation of bsTCE treatment. These effects were dependent on the presence of PBMCs. Several doses and dosing regimens were evaluated and were found to be effective.
In non-human primates (NHPs), the binding arms of the CD1d-Vδ2 bsTCE (CD1d VHH1D12, Vδ2-TCR VHH5C8) were not cross-reactive. To develop a cross-reactive bsTCE, the researchers substituted CD1d VHH1D12 with CD1d VHH1D22, which was cross-reactive to NHP CD1d, but bound to a different epitope on CD1d and did not activate type 1 NKT cells, eliminating the ability to assess effects on activation of NKT cells (which are significantly lower in abundance than Vγ9Vδ2-T cells). As no cross-reactive Vγ9- or Vδ2-TCR-specific VHHs were found, the researchers substituted Vδ2-TCR VHH5C8 with the scFv from the anti-Vγ9-TCR mAb, 7A5, which was shown to have reactivity with Vγ9 in one NHP model. Linking the 7A5 scFv to the CD1d VHH1D22 resulted in a CD1d-Vγ9 bsTCE that effectively bound to CD1d+ NHP monocytes and to NHP Vγ9Vδ2-T cells, triggering degranulation of Vγ9Vδ2-T cells.
In a single escalating dosing study, the CD1d-Vγ9 NHP-bsTCE had a short plasma half life, was rapidly cleared from plasma, and showed a large apparent volume of distribution. Dose-dependent binding to peripheral blood Vγ9-T cells was observed, and was more evident in the control Vγ9 bsTCE group, which could reflect the size of the antigen pool (CD1d and the Vγ9-TCR versus the Vγ9-TCR alone) and differences in Vγ9-T cell activation. In line with this, a transient decrease in peripheral blood Vγ9-T cells and upregulation of activation marker CD69 was observed following treatment with CD1d-Vγ9, but not with the control. Similarly, seven daily doses of CD1d-Vγ9 induced a moderate dose-dependent increase in several proinflammatory cytokines, including IL-2, TNF, IL-15, IL-6, and the chemokine CCL2, after the first administration. No weight loss nor bsTCE-related toxicities were observed, suggesting that CD1d-Vγ9 was well tolerated.
Together these results support the use of bsTCEs targeting CD1d and Vγ9Vδ2 TCRs to activate type 1 NKT cells and Vγ9Vδ2 T cells against CD1d+ cancer cells. Treatment showed promising antitumor efficacy in mouse models of MM, AML, and CLL, and was well tolerated in NHPs, supporting the initiation of a phase 1/2a study of a CD1d-Vδ2 bsTCE (LAVA-051) in patients with therapy-refractory CLL, MM, or AML.
Write-up and image by Lauren Hitchings



