
The ACIR team attended the CIMT Annual Meeting 2023 in Mainz, Germany. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
Tumor immune microenvironment and implications for immunotherapy
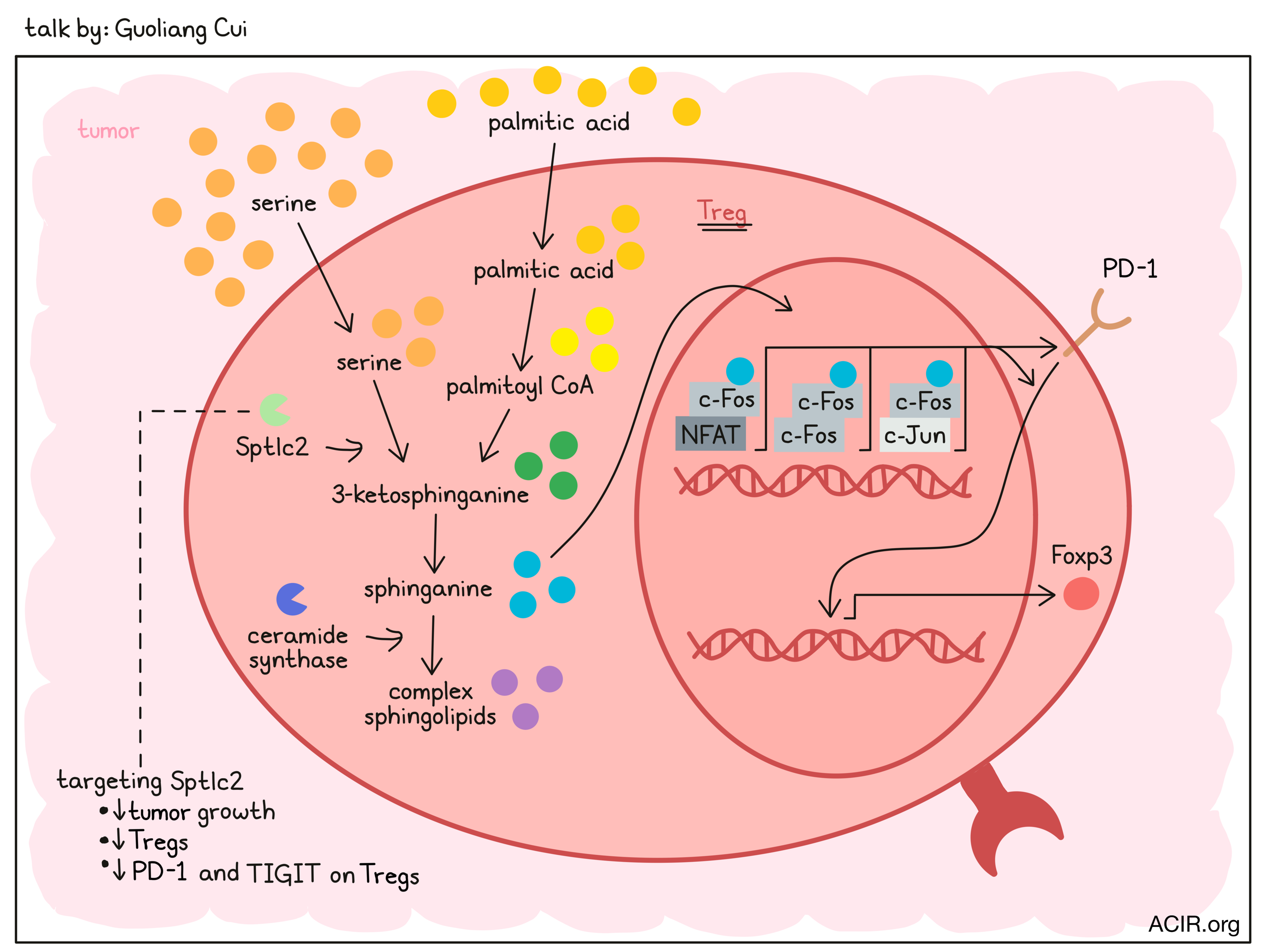
Guoliang Cui
Rafi Ahmed
Miriam Merad
Ira Mellman
Judit Díaz-Gómez
Naveen Mehta
T cell therapies and bispecifics
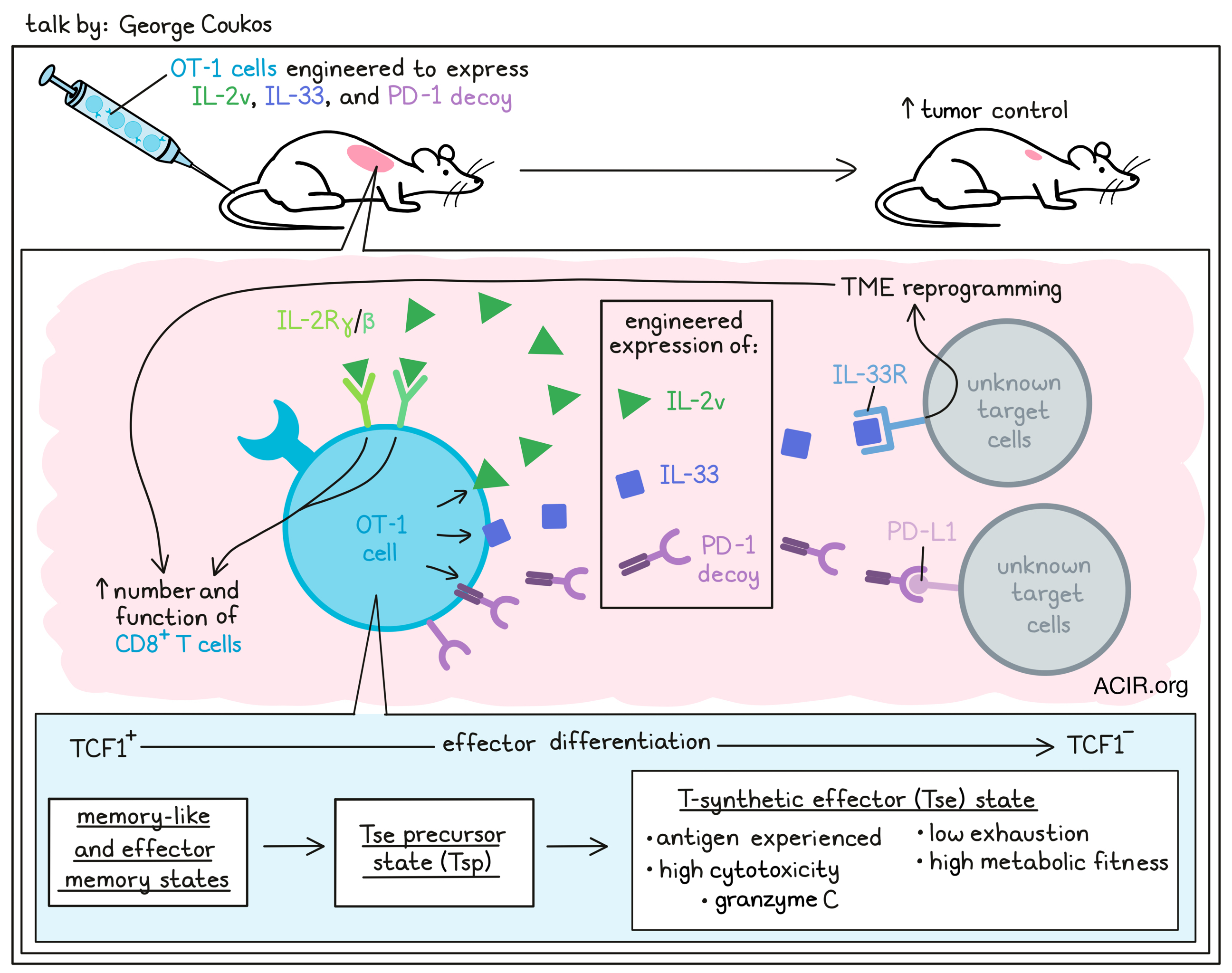
George Coukos
Kristen Vogt
Koustubh Ranade
Johanna Olweus
Targeting tumor antigens
Sebastian Amigorena
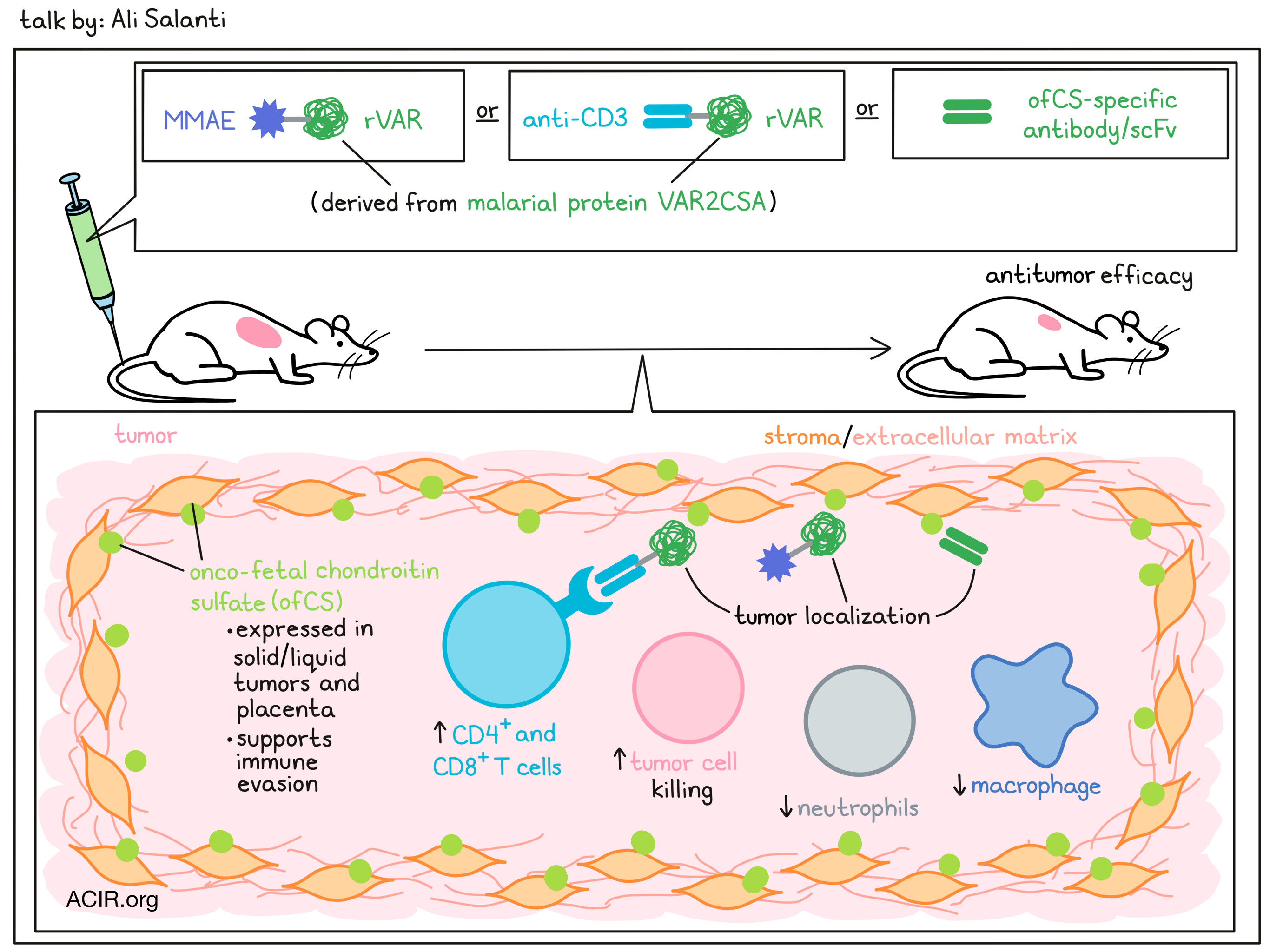
Ali Salanti
Niklas Grassl
Marij Welters
Federica Benvenuti
Vinod Balachandran
CIMT Lifetime achievement award
Hans-Georg Rammensee
In addition, we have two of our regular spotlights for you - so, keep scrolling!
Tumor immune microenvironment and implications for immunotherapy
Regulatory T-cell metabolism in the tumor microenvironment - Guoliang Cui - HI-TRON/German Cancer Research Center DKFZ, Mainz/Heidelberg, Germany
To systematically identify metabolic pathways that regulate Treg accumulation in the tumor microenvironment, Guoliang Cui and his team used a commercially available kit to discover 630 metabolites in the tumor and analyzed three different murine tumor types. In an unbiased computational approach, they identified the sphingolipid pathway to be important and found that substrates for de novo synthesis of sphingolipids, palmitic acid (the precursor to palmitoyl CoA) and serine, were enriched in the tumor microenvironment. In the first steps of this pathway, the enzyme serine palmitoyltransferase long chain base subunit 2 (Sptlc2) catalyzes the condensation of palmitoyl-CoA and serine to 3-ketosphinganine, which is then converted into sphinganine, and ultimately into more complex sphingolipids. Tumor-infiltrating Tregs had higher protein levels of the Sptlc2 compared to Tregs in spleens, suggesting that Tregs are able to integrate and respond to this change in substrates in the tumor microenvironment. Treg-specific ablation of Sptlc2 in SptlcFl/FlFoxp3YFP-Cre mice led to reduced tumor growth, a decrease in tumor-infiltrating Tregs, and reduced expression of PD-1 and TIGIT on Tregs in the tumor. In an elegant set of experiments, Cui and colleagues found that Treg-specific ablation of Sptlc2 only had an effect on tumor growth when serine was enriched in the tumor microenvironment. Blocking the further conversion of sphinganine to complex sphingolipids via inhibition of ceramide synthase indicated that the sphingolipid pathway was important for expression of both Foxp3 in CD4+ cells and PD-1 in Tregs. To understand the mechanism, Cui went on to show that the sphingolipid pathway metabolite sphinganine can directly bind to c-Fos protein and acts as a “molecular glue”, stabilizing c-Fos homodimers and c-Fos heterodimers with other transcriptional regulators (e.g., NFAT2 and c-Jun) and promoting the recruitment of c-Fos to chromatin-accessible regions of target genes, including genes encoding PD-1 and PD-1 signaling pathway components. Increased surface expression of PD-1 and PD-1 signaling then induces the expression of Foxp3 and Treg differentiation. In summary, Cui provided proof that increased serine levels in the tumor microenvironment promote Treg accumulation via metabolites of the sphingolipid pathway and c-Fos–PD-1 signaling, potentially providing additional approaches to eliminate tumor Treg cells.
T-cell lifestyle during chronic infection and cancer: implications for immunotherapy - Rafi Ahmed - Emory University School of Medicine, Atlanta, United States of America
In his keynote lecture, Rafi Ahmed summarized the research he pioneered on the phenotype, trajectory, and function of CD8+ T cells undergoing chronic antigen exposure, found initially in certain viral infections and later in cancer. In contrast to the priming response following initial infection, where highly cytolytic T cells rapidly rise (and fall) after viral exposure and typically fully control the infection, long-term viral persistence invokes mechanisms of differentiation leading to a “chronic resource cell” that preserves stem-like, undifferentiated features of mostly quiescent cells that retain the potential for proliferation and tissue-residence in lymphoid tissue. These stem-like CD8+ T cells are generated within the first weeks of an infection from naive T cells. At any given time, these cells slowly self-renew and differentiate into highly functional effectors, which exit the lymphoid tissues and transit through blood to target and kill the antigen-expressing cells, controlling the chronic infection and keeping the virus in check. Following PD-1 axis blockade, it is the release of these newly differentiated cells that is responsible for antiviral or antitumor immunity. This burst of proliferation and enhanced effector function is followed by rapid differentiation to a terminal, dying phenotype with low effector capability and retention in both target and lymphoid tissues. Accompanying these phenotypic changes are now well described changes in expression of cytokines/chemokines and their receptors, effector molecules, stimulatory and inhibitory immune modulatory molecules, and alterations in key canonical transcription factors (e.g., TCF1 and TOX). After reviewing the seminal studies in chronic viral infection and cancer, Ahmed presented new work showing (1) the presence of such stem-like PD-1+TCF1+CD8+ T cells in lymphoid-like structures in human HPV-positive head and neck cancer, (2) a highly selective TCR repertoire among the HPV-targeting cells (interestingly, T cells specific to the HPV E2 and E5 proteins and not the canonical E6 and E7 oncoproteins were most abundant, providing new opportunities for vaccines and TCR-engineered T cells) and (3) a replication of the phenotypic features of stemness and function. Finally, he described exciting results that demonstrate the improved quality of effector cells resulting from adding IL-2 (dependent on the high-affinity CD25 IL-2 receptor subunit) to anti-PD-1 axis therapy, producing effector cells with higher functional capacity, lower exhaustion signatures, and distinct transcriptional profiles from resource stem-like CD8+ T cells. These results showed that differentiation toward the terminal exhausted state is not immutable, and that there are opportunities for more effective therapies.
Targeting dendritic cells in cancer - Miriam Merad - Icahn School of Medicine at Mount Sinai, New York, United States of America
Inflammatory cells in the tumor immune microenvironment (TIME) direct disease outcome. To unravel and harness the mechanisms behind this, Miriam Merad focused on the roles of dendritic cells, which deliver immune cargo to lymph nodes to initiate T cell responses, and macrophages, which can both control inflammation to promote repair, and cause immunosuppression. Capitalizing on the presurgical setting, Merad described the advantages of the TARGET platform (The neoAdjuvant Research Group to Evaluate Therapeutics), which she and an integrated multidisciplinary team established to both evaluate the TIME at baseline and following immune perturbation. In already published work from the TARGET platform, a subset of PD-1hiIL-21+CXCL13+ Tfh-like CD4+ T cells and effector CD8+ T cells were expanded in patients with hepatocellular carcinoma responding to PD-1 blockade compared to non-responders, who had higher levels of terminally exhausted CD8+ T cells and Tregs. Given that tumor lesions of both responders and non-responders were highly infiltrated with T cells (present prior to therapy and expanded, but on different phenotypic trajectories following treatment), Merad investigated the local cues leading to different responses and turned to PIC-Seq – a technique that could identify the phenotypes of physically interacting cells isolated from tumor tissue. Interestingly, triple interaction between immunoregulatory mregDCs, CXCL13+CD4+ T cells, and progenitor CD8+ T cells was identified in responders and was correlated with enrichment of CD8+ effector cells, but was also present in non-responders. The mregDCs and CD4+ T cells shared multiple receptor–ligand pairs that led to effector CD8+ cells in responders, but terminal cells in non-responders, leading to the question of how mregDCs in the different niches differentially function and result in such different outcomes. Prior work had shown that uptake of cell debris induced the mregDC phenotype, which shut down further debris uptake and induced antigen presentation capability of the acquired cargo. While this shift was independent of multiple known migration-regulating molecules, mregDCs were present in normal and inflamed tissue and showed induction of cholesterol biosynthesis. Merad reported that DC maturation was dependent on movement of cholesterol to the cell surface, and that inhibition of cholesterol transport reduced DC maturation and the ability to stimulate CD8+ and CD4+ T cells. One highly significant effect of cholesterol transport inhibition was the reduction of IFNγR1 on the cell surface, and Merad tied this to other data indicating that aberrant glycosylation of IFNγR1 reduced the presence of IFNγR1 in lipid nanodomains on the surface and reduced IFNγR1 signaling. Similarly, inhibition of cholesterol movement reduced IFNγR1 in lipid nanodomains. Knockout of IFNγR1 in DCs significantly reduced IL-12 production and tumor control. Tying these results together, Merad suggested that it may be possible to modulate mregDC function by properly constituted lipid nanoparticles, thereby shifting the immune modulatory behavior of mregDCs to an antitumor state.
Mechanistic basis of cancer immunotherapy - Ira Mellman - Genentech, San Francisco, United States of America
The cancer immunity cycle defines the key steps in generation and maintenance of spontaneous immune responses to the tumor. It also serves as identification of points of breakdown of effective immunity requiring intervention to ideally correct. Ira Mellman used the cycle to set the stage for first describing the emerging understanding of how checkpoint inhibitors work, through T cells, to elicit antitumor responses, and then how T cells are profoundly influenced by the tumor immune microenvironment (TIME). Chronic antigen exposure, which occurs in tumors, has now been shown to result in the eventual production of “exhausted” T cells, marked by poor functionality and limited lifespans. Until recently, it was thought that anti-PD-1 axis therapy was reversing or preventing T cell exhaustion. The understanding of the epigenetics of exhaustion has made this explanation untenable, and now it is recognized that interruption of the inhibitory PD-1 axis functions earlier in the lifecycle of the T cell, leading to more de novo activation and improved differentiation. Elegant published in vitro and in vivo studies have shown that PD-1 signaling blocks critical CD28 costimulatory signaling and that PD-L1+ dendritic cells are critical for anti-PD-1 axis effects. Mellman then described more recent work demonstrating that TIGIT, a cis competitor of the costimulatory receptor CD266 for PVR (the activating ligand for CD226), prevents maximal T cell stimulation (both CD226 and CD28 are dephosphorylated by Shp2 recruitment to PD-1 immunoreceptor tyrosine-based inhibitory motif [ITIM]) independently from PD-1. This suggested that blockade of both pathways would be optimal. Combined PD-L1 and TIGIT blockade in patients with NSCLC showed promise in improving overall survival in a phase 2 study and is awaiting final results in a phase 3 study. Mechanistically, combination blockade improves the quality of TILs by dramatically upregulating CD226 and downregulating TOX in both antigen-specific and bulk CD8+ T cells. The decrease in TOX expression requires CD226 signaling. A very large single-cell analysis clearly described multiple T cell states and key markers along the continuum from naive to dying cells, including Slamf6-marked T stem cell memory (TSCM) cells, interferon pathway-marked effector cells, CCL5-marked transitory effector-like cells, and a cluster of late-stage effector/exhausted cells. TCR sequencing analysis demonstrated that the combined PD-L1 and TIGIT blockade led to a dramatic expansion of antigen-specific T cells in the draining lymph nodes (dLNs), blood, and tumor. Pseudotime analysis indicated that although individual checkpoint therapy led to improvements (such as an increase of effectors in the dLN, trafficking of effectors to tumors from dLNs, reduction in the exhausted compartment), combination therapy optimized multiple effects, leading to expansion of TSCM and T effector-like cells in the dLN and trafficking of polyfunctional, oligoclonal cells through the blood and to the tumor. In the tumor, effector memory and CTLs appear, with more limited final differentiation to the terminally exhausted phenotype. Limited differentiation to exhaustion appears to be dependent on co-stimulatory pathways. Preliminary data suggests that the Fc domain on the TIGIT antibody may signal myeloid cells to secrete proinflammatory factors, limit Treg suppression, and activate NK cells, suggesting multiple pathways may be active, which is supported by biomarker data from a phase 2 combination trial. Finally, Mellman described a novel high-throughput assay (soon to be published) creating an array of “wells” in mouse ears that can be seeded with tumor cells. The growth of the cells can be independently monitored in each well, and perturbations such as checkpoint blockade or growth factor addition can be tested. Different tumor cell lines showed different behavior upon implantation, indicating a tumor cell autonomous effect. Importantly, even when seeded with homogeneous tumor cell samples, tumor cell growth and T cell infiltration was variable well-to-well, indicating an impact of the local cellular environment on tumor growth, despite exposure to a similar T cell response.
Unravelling tumor-intrinsic resistance mechanisms to T-cell mediated cytotoxicity in pancreatic cancers - Judit Díaz-Gómez - Vall d'Hebron Institute of Oncology (VHIO), Barcelona, Spain
To explore new avenues that may render pancreatic cancer susceptible to immunotherapy, Judit Diaz-Gomez and colleagues performed a genome-wide CRISPR/Cas9 knockout screen to discover genes that limit the in vitro killing of a patient-derived pancreatic cancer cell line, TCL-4177, by neoantigen-specific TCR-transduced T cells. Focusing on genes that enhanced the susceptibility to T cell killing when knocked out, the researchers identified 35 genes that restrain T cell mediated killing and have not been previously studied. Plotting these genes in a proteome-wide analysis network revealed involvement of the autophagy and ubiquitination pathway, chromatin remodeling, vesicle trafficking, protein folding, and ESCRT complex in tumor intrinsic resistance mechanisms. So far, seven out of 10 genes involved in four different pathways were individually validated, and silencing of four of these genes increased killing in another pancreatic tumor cell line, TCL-CABA, by T cells enriched for a different (unknown) specificity. The most promising candidates, ITGB1 and MARCH5, were further analyzed retrospectively in clinical data, which demonstrated that low expression of ITGB1 or MARCH5 correlated with improved overall survival in patients with pancreatic ductal adenocarcinoma. Further analysis and validation of additional candidates, as well as mechanistic studies are currently ongoing.
CLN-617 is a first-in-class fusion protein that retains IL-2 and IL-12 in injected tumors and potently triggers systemic anti-tumor immunity - Naveen Mehta - Cullinan Oncology, Cambridge, United States of America
Multiple lines of evidence indicate that IL-2 and IL-12 are not only potent antitumor cytokines and orthogonal in signaling pathways, but also synergistic in activity. However, each alone is hindered by toxicities when delivered systemically in the clinic. To overcome such limitations, CLN-617 was designed based on three principles: (1) cytokines mainly act in an autocrine/paracrine manner and should be delivered locally; (2) local injection of a cytokine requires modifications to limit systemic exposure and enhance retention; and (3) cytokines often act in concert with other cytokines. Thus, a single molecule was designed to encode unmodified human (h)IL-2 and hIL-12 linked to a collagen binding domain (hLAIR2) and human serum albumin to retain injected CLN-617 in the tumor. CLN-617 bound collagen with single-digit nM affinity, and retained full IL-2 and IL-12 stimulatory activity in in vitro assays. Intratumoral injection of a murine version of CLN-617 (mCLN-617) led to intratumoral concentrations that were more than 10x higher than the maximal concentration observed systemically. In multiple tumor models, including models resistant to anti-PD-1 axis therapy, monotherapy improved survival following a single intratumoral (i.t.) injection. For example, in MC38, 10/10 complete responses (CRs) were observed with i.t. mCLN-617 versus 0/10 CRs with systemic anti-PD-1. Therapy was also well tolerated. Rechallenge experiments in surviving mice 60 days after initial tumor implantation demonstrated effective, potent memory responses. Further, subcutaneous dual flank and liver metastasis models demonstrated potent abscopal effects, especially in conjunction with anti-PD-1 therapy. Importantly, analysis of both injected and non-injected tumors demonstrated an enhanced CD8+:Treg ratio, and a significant induction of tumor-specific CD8+ T cells was observed in the blood, demonstrating systemic effects. A clinical study is targeted to begin in 2023 in conjunction with PD-1 axis blockade.
T cell therapies and bispecifics
Learning from natural immunity to create synthetic immunity - George Coukos - CHUV/UNIL-Ludwig Institute for Cancer Research, Lausanne, Switzerland
Adoptive cell therapy (ACT) with tumor-infiltrating lymphocytes (TILs) is a well described form of immunotherapy that can lead to significant and durable responses and a very hopeful prognosis in patients who experience a complete response. George Coukos described the establishment of a TIL therapy scheme for patients with melanoma in Lausanne, Switzerland, in which patient TILs were expanded ex vivo using a rapid expansion protocol and infused back to the patient following non-myeloablative chemotherapy. IL-2 was used to support in vivo expansion of the transferred TILs, followed 30 days later with anti-PD-1 therapy every two weeks. Thirteen patients with melanoma – all of whom had been treated with prior anti-PD-1 axis therapy and were either refractory, developed resistance or stopped due to toxicity – were enrolled and received TILs. Six patients were clinical responders, while the remaining seven were non-responders, which replicated the experience at other institutions. A set of baseline samples for detailed analysis of the tumor microenvironment (TME) before therapy was available for all patients. Single-cell sequencing, confirmed with multiplex immunofluorescence microscopy (mIF), showed that responders had more inflamed, T cell-rich tumors, and that the infiltrating T cells exhibited an exhausted, TCR-signaling, antigen-experienced (PD-1+), and cytotoxic phenotype with markers of CD28 signaling. To take further advantage of such relevant cells, the Neo-TIL process was developed, consisting of identifying patient-specific neoantigen peptides and using these to pulse autologous APCs to specifically expand neoantigen-specific T cells for 10 days prior to nonspecific T cell expansion. Some tumor-associated antigens, like MAGEA10, WT-1, and HER2, were also used. This process was very effective across multiple peptides and multiple tumor histologies, resulting in 10–100x increases in frequency of tumor-specific CD8+ T cells, sometimes constituting 75% of the product. Three patients were anecdotally described following treatment with 50-70 billion expanded Neo-TILs (which were 17-50% tumor-specific). One of these patients achieved a complete response. The second saw a rapid partial response (PR), followed by progressive disease with an immune-edited phenotype (loss of HLA expression). The third patient, whose tumor had previously regressed following TIL therapy, experienced a PR marked by massive new T cell infiltration, but the tumor eventually escaped with an immune-edited phenotype. Analysis of TILs from the original tumor showed extensive and “noisy” cross-talk between multiple cell types in responding tumors, while showing a pattern devoid of interactions in non-responding tumors. Multiplex immunofluorescence microscopy was used to investigate cell “neighborhoods” and revealed a strong presence of closely interacting CD8+PD1+ T cells and CD11c+ DCs in responding patients (both in the tumor and surrounding stromal tissue), suggesting these are the nexus of generating an effective TIL product. Furthermore, following up on the presence of immunological synapses detected by mIF, CYTOF analysis showed that CD28-expressing TILs had a stronger effector and effector precursor signature, and were particularly rich in signals of IL-2 signaling. This observation led to the production of gene-engineered OT-1 T cells expressing an IL-2 variant (specific for the lower affinity IL-2 receptor βγ), the alarmin IL-33, and a PD-1 decoy (which eventually was shown to be irrelevant to activity). In just-published studies, these T cells differentiated into a novel “synthetic effector” state, which was antigen-experienced (but not exhausted), highly cytotoxic (especially marked by expression of Gmzc) and showed superior metabolic fitness. In a cold tumor, the engineered T cells proved to be very effective and could be recovered from the TME 12 days after infusion. Importantly, the cells were effective without lymphodepletion or supportive IL-2.
Tumor microenvironment actuated GD2 CAR T cells prevent on-target off-tumor toxicities - Kristen Vogt - Memorial Sloan Kettering Cancer Center, New York United States of America
Finding highly specific targets for CAR T cell therapy is a challenge, and high expression of potentially desirable targets in normal tissues can result in significant on-target, off-tumor toxicities. GD2 is a prime example, demonstrating very high expression in various brain tissues. To overcome this problem, boolean AND-gated logic circuits have been developed for CAR T cells, requiring activation by one tumor-specific target in order to induce expression of the CAR targeting a second target to initiate cytotoxicity. The synNotch circuit, which releases a transcription factor, is one such circuit. Kristen Vogt hypothesized that activation of the circuit could be achieved with a tumor microenvironment (TME)-specific marker found on normal cells in the TME to activate CAR expression for a tumor cell-specific target, restricting cytotoxicity to the tumor bed. P-selectin is overexpressed on the vasculature of solid tumors and in response to radiation, and so was inserted as the actuating target in synNotch CAR T cells directed to CD19 as the tumor-specific target. Recombinant P-selectin, P-selectin expressed in cis on the target cell, or P-selectin expressed in trans on non-target cells were all able to activate the circuit and result in CD19-targeted cell killing. This approach was then applied to the GD2 CAR system. Notably, GD2 is a desirable target for many solid tumors, but high-affinity GD2 CARs are known to demonstrate fatal neurotoxicity due to the high expression in normal brain tissue. P-selectin-gating of GD2 CAR T cells significantly reduced infiltration into the brain and associated neurotoxicity. In a P-selectin+GD2+ K562 subcutaneous tumor model, intravenously delivered P-selectin-gated CAR T cells were able to control tumor growth, and demonstrated no infiltration into the brain.
Tebentafusp-a gp100 directed TCR-CD3 bispecific for the treatment of metastatic uveal melanoma - Koustubh Ranade - Immunocore, Rockville, United States of America
Tebentafusp (Kimmtrak®), a first-in-class, off-the-shelf, bispecific protein consisting of an anti-CD3 scFv and a soluble affinity-enhanced T cell receptor targeting the gp100 (YLEPGPVTA)-HLA*A02:01 complex has recently received approval in 30+ countries for the treatment of patients with metastatic uveal melanoma (mUM). In contrast to cutaneous melanoma, uveal melanoma has a low tumor mutational burden and low responsiveness to checkpoint blockade. According to Koustubh Ranade, up to 50% of patients with uveal melanoma develop metastatic disease, with the liver being the primary site of metastasis. In a randomized phase 3 clinical trial in previously untreated HLA*A02:01-positive patients with mUM, treatment with tebentafusp resulted in a median overall survival of 21.7 months compared to 16.0 months for investigator’s choice of therapy (82% treated with single-agent pembrolizumab, 13% with ipilimumab, or 6% with dacarbazine). Survival curves separated as early as 1 month from start of treatment, and Ranade indicated that in data to be released soon, the survival curve for patients treated with tebentafusp ended in a long tail. Adverse reactions (ARs) were predictable, manageable, and consistent with the mode of action (e.g., cytokine release syndrome and rash). The majority of ARs appeared within the first 3 weeks. In tumor biopsies taken at day 16 after three doses of tebentafusp, the tumor microenvironment was heavily infiltrated by T cells, and type 1 and 2 interferon pathways were highly induced. Ranade also presented another bispecific TCR-TCE that is currently in the pipeline and targets the cancer testis antigen PRAME, which is expressed in various solid tumors in complex with HLA*A02:01. In a phase 1 dose-escalation study, this bispecific TCR-TCE has shown encouraging clinical activity in multiple tumor types.
Exploring T-cell receptors to access new categories of cancer immunotherapy targets - Johanna Olweus - Oslo University Hospital, Oslo, Norway
Johanna Olweus, substituting for Marcela Maus, focused on the advantages and classes of TCR-based T cell therapies, which, by recognizing MHC-presented peptides from all cellular compartments, have a demonstrably broader set of potential targets than CAR T cells. Most trials using TCR-engineered T cells to date have targeted TAAs (both cancer testis and oncofetal antigens), which are shared across multiple patients, but often demonstrate heterogeneous and low expression. Clinical success targeting TAAs has been limited, although recent trials targeting MAGE-A4 or WT1 have shown indications of clinical impact. Individual personal neoantigens generated by single-nucleotide variants have also been targeted. Although neoantigens are highly tumor specific and have the potential to be highly immunogenic, typically only a very low proportion of neoantigens are immunogenic. In a clinical trial capturing up to 3 neoantigen-specific TCRs per patient isolated from autologous T cells, minimal clinical effect was observed, with stable disease as best response in 5 of 16 patients. The impact may have been affected by the low affinity of these TCRs. Viral onco-antigens represent a highly and homogeneously expressed target in virus-driven cancers, and T cells engineered with a TCR with high affinity to an HPV-E7 epitope showed partial responses in half of the 12 treated patients. A newer target class are cell-type specific targets, capitalizing on unique biology. As an example, Olweus described her work targeting terminal deoxynucleotidyl transferase, an enzyme critical to early differentiation of T and B cells, but not expressed in mature circulating cells. This transient expression in normal cells, but constitutive overexpression in various hematopoietic malignancies opens a window of opportunity for an engineered TCR-T cell therapy. Using high-affinity TCRs targeting TdT, Olweus’s group showed high tumor specificity, sparing normal T and B cells, and thymopoiesis and hematopoiesis in vitro and in humanized mice. Shared, onco-protein neoantigens, rather than personal neoantigens, represent another potential target, although few (~1%) generate spontaneous responses in patients. Healthy donors can be a source of TCRs to this “neglected” antigen class, and Olweus described isolation of a high-affinity TCR targeting a mutation in the cytokine receptor Flt3, which is prevalent in AML. Finally, since tumor mutation burden and spontaneous T cell infiltration do not always correlate, tumor antigens that are not genetically encoded may play a role. Olweus described two such antigen classes stemming from epigenetic dysregulated splicing and translation mistakes, which may provide more promising TCR-engineered T cell therapy opportunities. Wrapping up, Olweus emphasized the importance of high and homogenous antigen expression, and identification of high-affinity TCRs.
Targeting tumor antigens
Epigenetics tumor antigens - Sebastian Amigorena - Institut Curie, Paris, France
To enhance the library of useful tumor antigens, Sebastian Amigorena and his team investigated the “dark genome” - genome segments typically not transcribed, but aberrantly expressed in tumor cells due to dysregulation. In particular, he focused on finding novel antigens resulting from the combination of cancer-driven derepression of epigenetically silenced transposable elements (TEs), which are broadly spread throughout the genome near and within canonical protein-coding genes, and widespread alterations in splicing machinery found in tumor cells. Derepression of TEs and novel splicing can result in aberrantly spliced fusion products (“JETs”; Junctions between Exons and Transposable elements) containing novel fusion antigens. Amigorena’s prior work described a bioinformatic pipeline that searched for such novel transcripts in murine tumor cell lines, predicting multiple candidate antigens, which showed tumor and tissue of origin specificity. Mass spectrometry (MS)-based immunopeptidomics detected cognate peptides, some of which were shared among tumors, and many of which were immunogenic in tumor-bearing animals and could also control tumors following prophylactic vaccination. Knockout of the epigenetic regulator Setbp1 confirmed the epigenetic origin of the JETs and delayed tumor growth in the B16-OVA model. Anti-PD-L1 therapy significantly enhanced this effect and the induction of JET-specific T cell responses. In newer work, a similar pipeline applied to human tumor samples also identified tissue- and tumor-specific JET expression. For 600 lung cancer samples, ubiquitous, tumor-specific, and tumor-associated JETs could be identified, many with low expression in juxta-tumor samples or in the normal tissue GTEX database. Stringent MS immunopeptidomics of 17 fresh lung tumor specimens supported presentation of multiple peptides by HLA class I. Analysis of corresponding TILs detected multiple CD8+ T cells directly ex vivo (for 5 of 32 tested peptides), and more following peptide-stimulated in vitro expansion (for 19 of 32). Amigorena wrapped up by describing the collaboration with MNEMO Therapeutics to identify scFvs targeting human JET peptides presented by cognate HLAs to develop T cell-engaging bispecifics and CAR T cells. Encouraging data has demonstrated super-high-resolution immunofluorescence detection on naturally presenting cell lines and in vitro and in vivo efficacy.
Targeting onco-fetal glycosaminoglycans omnipresent in the tumor microenvironment - Ali Salanti - University of Copenhagen, Copenhagen, Denmark
Ali Salanti, a malaria researcher, made an interesting discovery when he recognized that plasmodium falciparum can engineer infected erythrocytes to express a malarial-encoded cell surface protein, VAR2CSA, which binds a distinct type of chondroitin sulfate (CS), exclusively expressed in the placenta during fetal development, providing the parasite with a new niche to avoid circulation and subsequent clearance in the spleen. The distinct CS pattern found in the placenta allows the formation of a reservoir for cytokines; rapid, invasive growth; and immune evasion of the trophoblast. Similar CS features characterize malignant cells, and recombinant VAR2CSA (rVAR2) was shown to bind to virtually all tumor cell types, but minimally or not at all to normal cells (except in the placenta), identifying the complex secondary CS modification as an onco-fetal tumor antigen (ofCS). Intravenously injected rVAR2 localized to the tumor in mice and killed tumor cells when conjugated with the microtubulin inhibitor MMAE. In an effort to minimize immunogenicity of the malarial protein and expand the half-life of such a treatment, Salanti focused on developing an antibody specific for ofCS. This turned out to be extremely challenging in many species, but Salanti and team eventually screened large phage display libraries and identified two monoclonal antibodies that bind with high affinity and specificity to both human and murine ofCS. Higher resolution confirmed binding of ofCS scFv to stroma and extracellular matrix in malignant tissue, but not normal tissue with high amounts of CS. Furthermore, not only solid tumors, but also hematological tumors were found to express ofCS, and Salanti showed binding of anti-ofCS to human AML, but not normal bone marrow. In KpC mice, which are genetically engineered to develop pancreatic cancer, ofCS scFvs stained the very early onsets of tumor-initiating cells. To elucidate the role of ofCS in tumor development, Salanti and team knocked out an enzyme that is needed to create ofCS. Tumors with this knockout were more heavily infiltrated by CD3+ cells, and subset analyses revealed a dramatic shift from infiltrating neutrophils and macrophages to CD4+ and CD8+ T cells and reduced tumor growth, suggesting a role of ofCS in tumor immune evasion. The anti-ofCS scFvs demonstrated in vivo tumor localization and anti-tumor activity when conjugated to MMAE or to an anti-CD3 scFv (to create a T-cell engaging bispecific), including generation of immune memory. No weight loss was observed. A phase 0 study has been planned to inform dosing and pharmacokinetic for the following phase 1 trial.
A long peptide vaccine targeting the clonal driver mutation H3K27M in adult patients with diffuse midline glioma - Niklas Grassl - German Cancer Research Center DKFZ, Heidelberg, Germany
Diffuse midline glioma (DMG) predominantly occurs in children and young adults, and has a grim overall survival of just about 9 months. Histone H3 K27M is a recurrent clonal driver mutation in DMG, and a long H3K27M peptide vaccine reduced growth of subcutaneously transplanted H3K27M+ tumors in MHC-humanized mice. Niklas Grassl presented data from a first-in-human trial of a H3K27M long-peptide vaccine in 8 young adult patients with progressive DMG; 4 of the patients also received concomitant anti-PD-1 therapy. Treatment was well tolerated, with injection site reactions being the only vaccination-related adverse events. Following vaccination, H3 K27M-specific immune responses were observed in 5 out of 8 patients, with specificity toward the mutant peptide; no association with HLA type was observed. Overall survival was an encouraging 12.8 months, with a rapid transient tumor regression in 4 of 5 patients who showed vaccine-induced T cell responses, one of whom showed early pseudoprogression and is still alive after over 30 months. A proximity ligation assay and anti-MHC-II blockade of a peptide stimulation assay indicated that CD4+ T cells recognizing HLA-DR mediated the vaccine response. Paired αβ TCR cloning of mutant peptide-expanded T cells from peripheral blood of the long-surviving patient identified multiple antigen-reactive clonotypes and a marked increase in the frequency of many of CD4+ T cells bearing these expanded clonotypes in peripheral blood and cerebrospinal fluid of the patient following vaccination. Finally, Grassl indicated that a multi-center trial is now ongoing that combines H3 K27M peptide vaccine, radiotherapy, and an anti-PD-L1 antibody in newly diagnosed H3-mutated glioma patients.
Phase I/II clinical trial targeting alternative shared neoantigens (TEIPP) in checkpoint-resistant non-small cell lung cancer - Marij Welters - Leiden University Medical Center, Leiden, The Netherlands
Searching for new sources of shared antigens to be targeted in immunotherapy-resistant NSCLC, Marij Welters described an approach to turn an escape mechanism into a vulnerability. Tumors evade the immune system in multiple ways, but genetic instability of tumors and strong immune pressure against tumors can often result in reduced antigen presentation, which frequently involves downregulation of the transporter associated with antigen presentation (TAP), preventing MHC presentation of canonical epitopes. However, absence of the canonical TAP-dependent pathway allows a number of other peptides present in the ER to effectively compete and be bound to MHC. These are known as T cell epitopes associated with impaired peptide processing (TEIPP). TEIPP are derived from the non-mutated proteome, but have escaped central tolerance, and so may be highly immunogenic shared “neoantigens”, useful to target some escaped tumors. A pipeline was built for NSCLC samples to predict MHC binding of N-terminal signal and C-terminal tail peptides (known enriched sources of TEIPP peptides), analyze the immunopeptidome of TAP-deficient tumor cells and normal healthy tissue, and identiy reactive T cells and TCRs. An encouraging peptide derived from LRPAP1 was previously identified in this class and was highly immunogenic across multiple donors in vitro as a synthetic long peptide (SLP) vaccine after making a heteroclitic anchor residue (S>V) change. Welters then described the results of a recent clinical vaccination study in patients with NSCLC who relapsed after checkpoint blockade therapy. Patients were vaccinated with the SLP in adjuvant Montanide 3 times at 3-week intervals. At the first two dose levels, safety was established. Encouraging clinical responses were observed in 7 of 12 vaccinated patients, and LRPAP1-specific polyfunctional CD8+ and CD4+ T cells were detected using dextramer staining and intracellular cytokine staining of peripheral blood. Knockout of TAP1 in a NSCLC tumor cell line generated engineered tumor cells that could be recognized by sorted CD8+Dextramer+ PBMCs from a vaccinated patient, supporting TEIPP vaccination as a potential salvage therapy for immune-escaped tumors with impaired antigen processing machinery.
Type 1 DCs drive anti-tumoral immunity to endogenous immunogenic neoantigens in lung cancer - Federica Benvenuti - International Centre for Genetic Engineering and Biotechnology, Trieste, Italy
Focusing on the role type 1 dendritic cells play in selecting and generating effective CD8+ T cell responses to tumor neoantigens, Federica Benvenuti began by describing how knockout of Mlh1, a key enzyme in the DNA repair pathway, in epitope-poor KPcontrol (rasG12D/Trp53loss) cells created a novel cell line (KPneo) expressing non-surrogate, bona-fide neoantigens. 26 new novel epitopes were identified in KPneo, and implantation of KPneo in immunocompetent mice resulted in CD8+ T cell-dependent delay of tumor growth relative to the parental KP tumor cell line, and a massive increase in intratumoral CD8+IFNγ+ T cells. Multiple neoepitopes generated T cell responses. Analysis of cDC1s in implanted tumors showed a significantly higher number of cDC1s in KPneo compared to KPcontrol tumors. Deletion of the cDC1 compartment (Batf3 KO) showed that the effect of the neoantigens on T cell number and phenotype was totally dependent on the presence of cDC1s. Direct intratumoral injection of Flt3-ligand and polyI:C rendered KPneo, but not KPcontrol tumors sensitive to anti-PD-L1 treatment. This effect required cDC1s, based on results obtained with XCR1DTA mice. Analysis of human NSCLC data showed that both a cDC1 signature and CD8 effector signature were higher in TMBhigh versus TMBlow patients, supporting the observations in KPneo and KPcontrol mice. Orthotopically implanted KPneo tumors in the lung responded weakly to anti-PD-L1 therapy, but were significantly inhibited in growth by DC stimulation with Flt3L and agonist CD40 antibody (Flt3L/agOX40), and responses to the KPneo neoantigens were dramatically increased. Experiments in reporter mice expressing XCR1-venus showed a parallel increase in infiltrating cDC1s and CD8+ T cells in KPneo tumors under therapy. Single-cell analysis of CD8+ T cells in untreated versus Flt3L/agOX40-treated tumors showed differences in subset frequencies, and increased cytotoxic capacity and proliferation in treated tumors. Analysis of the cDC compartment showed a dramatic loss of mregDCs and an overall shift of the cDC1 and cDC2 compartments to a less exhausted/tolerogenic state with treatment, altogether suggesting that the DC compartment within tumors has a significant impact on both the repertoire of responding CD8+ T cells and the quality of their antitumor function.
Pancreatic cancer – clinical discovery to new immunotherapies - Vinod Balachandran - Memorial Sloan Kettering Cancer Center, Charlestown, United States of America
Vinod Balachandran studies pancreatic ductal adenocarcinoma (PDAC), which is the quintessential example of an immunologically cold tumor, with a low mutation burden, minimal T cell infiltration, and resistance to immunotherapy. However, around 9% of PDAC tumors are hot, and patients with such tumors have better survival. With the objective of identifying neoantigens that could most effectively prime de novo responses, amplify pre-existing responses, or both, Balachandran and colleagues developed a model for evaluating the quality of neoantigens by estimating “selfness” and “non-selfness” based on predicted MHC binding, potential TCR reactivity, and similarity to known viral antigenic peptides. Interestingly, the quality of neoantigens was better at predicting patient survival than the quantity of neoantigens, and T cells specific for high quality neoantigens could be identified in the blood of a number of long-term survivors of pancreatic cancer up to a decade later. The team also found that high quality neoantigens were likely to be edited in recurring tumors of long-term survivors, and that the spontaneous development of high quality neoantigens could lead to increased neoantigen-specific T cells that may delay tumor recurrence. Balachandran and colleagues wondered whether RNA vaccines could be used to spark de novo responses, and in a collaborative study, they developed individualized mRNA vaccines for pancreatic cancer. In a phase I trial, patients underwent surgery, and were later treated with atezolizumab at week 6, followed by 8 consecutive doses of a personalized cancer vaccine starting at week 9, and then standard-of-care chemotherapy starting at week 21, with a final late booster at week 46. While the trial was disrupted by the COVID-19 pandemic, 16 out of 19 patients who received atezolizumab went on to receive their vaccines, supporting the feasibility of the generation and administration of a custom vaccine after surgery. Primary ELISpot analysis and confirmatory TCR sequencing CloneTrack tests were used to monitor for vaccine-induced T cell responses, and together revealed robust post-vaccination responses that could be maintained even after chemotherapy. Clustering clones based on vaccine-induced clonal trajectories in peripheral blood showed that the polyclonal T cell pool expanded by the mRNA vaccines included clones that were undetectable prior to vaccination, and was distinct from the pool expanded by anti-PD-L1 – an effect that was particularly apparent after vaccine boosters, which further expanded the vaccine-induced, but not PD-L1-induced clones. Further, the researchers saw evidence of priming of de novo responses, including neoantigen-specific responses. Patients who showed both primary ELISpot activity and confirmatory clonal expansion were classified as mRNA neoantigen vaccine responders. Overall, mRNA vaccines induced T cell responses in 50% of PDAC patients, many of which were substantial and durable. At a median follow-up of 18 months, responders had not yet reached a median recurrence-free survival, while non-responders had a median recurrence-free survival of 13.4 months. This improvement was likely attributed to the vaccine. Considering that non-responders may be less fit for mRNA vaccination, the researchers compared patient responses to the COVID-19 vaccine, and found that cancer vaccine responders and non-responders mounted similar responses to the COVID-19 vaccine, ruling out baseline immune fitness as a contributing variable. They did, however, find that responders had more clonal mutations and higher-quality neoantigens. In a clinical vignette, Balachandran described a patient who appeared to have evidence of metastasis, along with a suspicious liver lesion, which ended up being a lymphoid aggregate of vaccine-expanded T cell clones that disappeared with later imaging. This result may represent vaccine-induced immune control of a micrometastasis. Moving forward, Balachandran and colleagues will be conducting a phase 2 clinical trial of this vaccine strategy, and are considering ways to scale this treatment to other checkpoint blockade-resistant cancers.
CIMT Lifetime Achievement Award
Hans-Georg Rammensee - University of Tübingen, Tübingen, Germany
Hans-Georg Rammensee received the CIMT Lifetime Achievement Award for his contributions to the field of cancer immunotherapy. Rammensee identified and described the rules that guide the interaction between peptides and MHC molecules, and with this, contributed significantly to the understanding of the specificity of T cell recognition, the establishment of T cell tolerance, and the design and function of vaccines. His use of mass spectrometry and bioinformatics, and the first exact predictions of naturally presented peptides laid the foundation for the development of epitope prediction tools, new technologies for T cell analysis, and the building of databases for MHC-presented peptides in health and disease. Further, Rammensee drove the translation of this knowledge towards the design of personalized therapeutic peptide and mRNA cancer vaccines. Seven companies have spun out of his research, including Immatics Biotechnologies and CureVac.
By Ute Burkhardt, Ed Fritsch, and Lauren Hitchings



