In an effort to investigate epigenetic mechanisms that contribute to tumor immune evasion, Liu, Liang, Xu, Dong, and Dong et al. recently explored the connection between the RNA demethylase FTO, increased glycolysis in tumor cells, and reduced T cell activation in the tumor microenvironment. Further, the researchers developed and tested a potential strategy for targeting this pathway to enhance immunotherapy. Their results were recently published in Cell Metabolism.
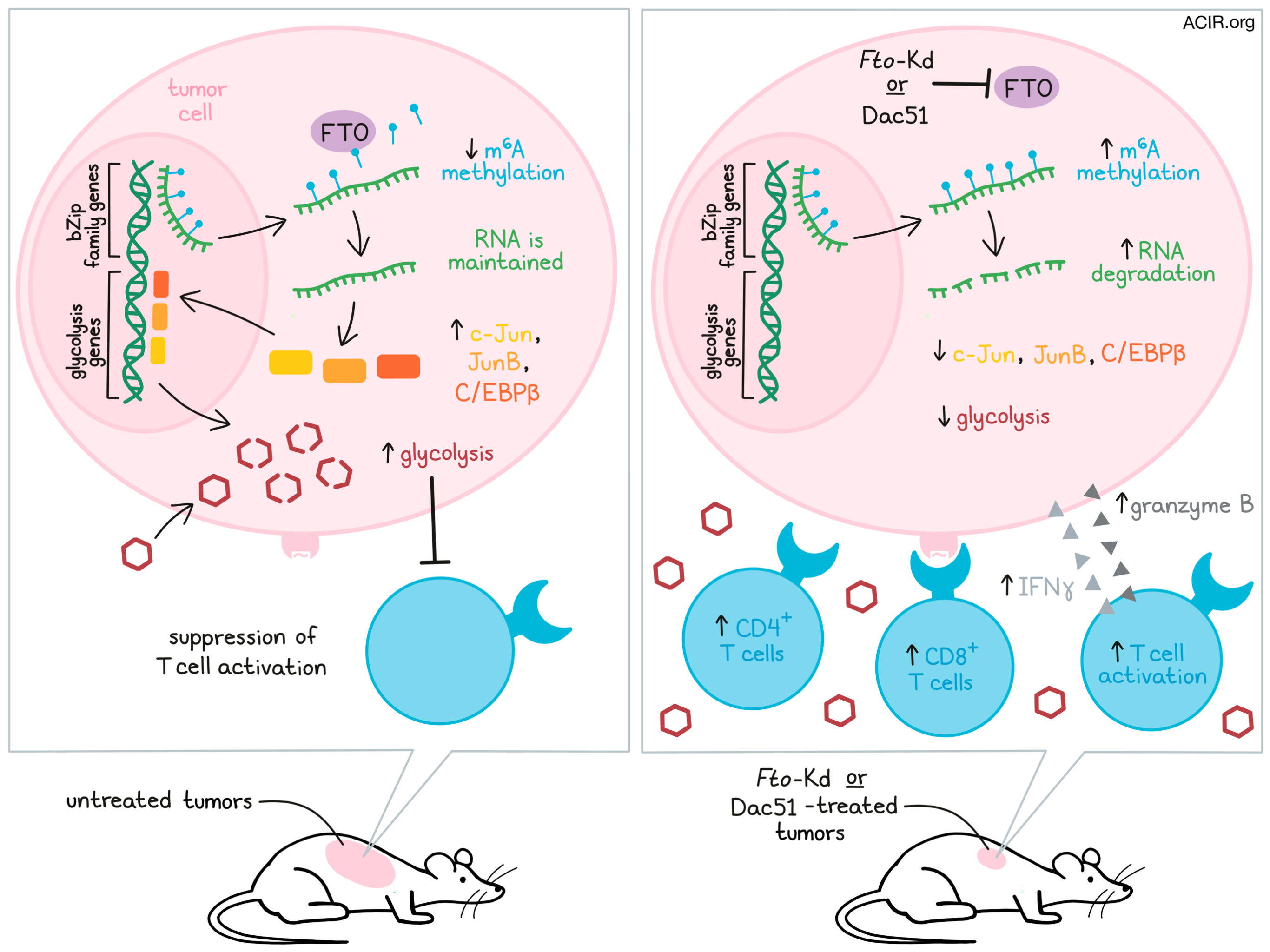
To begin, Liu, Liang, Xu, Dong, and Dong et al. analyzed bulk RNAseq data from TCGA and defined a cytolytic T lymphocyte (CTL) score that was associated with better patient survival. This CTL score was then found to negatively correlate with the RNA demethylase FTO (which demethylates m6A target sites in RNA, resulting in enhanced RNA degradation) in a wide variety of tumor settings. To directly determine the role of FTO, the team used shRNA to knock down Fto (Fto-Kd), and the efficacy of the knockdown was validated with observations of increased m6A methylation. In vitro and in immunodeficient mice, Fto-Kd and control tumor cell lines grew at similar rates, but in immunocompetent mice, Fto-Kd tumors grew more slowly, suggesting enhanced antitumor immunity. Looking more closely at the immune microenvironment, the researchers identified increased infiltration of CD4+ and CD8+ T cells, including tumor antigen-specific CD8+ T cells, into tumor tissues. In co-cultures, these T cells were also more activated and more functional, producing higher levels of IFNγ and granzyme B than T cells in control cultures, and were more effective at killing target tumor cells.
Interested in what might be causing this enhanced antitumor immunity, the researchers (after ruling out enhanced priming by dendritic cells) investigated whether FTO plays a role in glycolysis, which is utilized in tumors both for rapid cell growth and for inducing an immunosuppressive microenvironment that metabolically restricts effector T cell functions. In Fto-Kd cells, they found that glycolytic capacity was reduced, as were levels of metabolites of glycolysis pathways and the release of glycolytic byproducts. As expected, Fto-Kd also enhanced multiple markers of T cell activation. Administration of oligomycin to enhance the glycolytic capacity of tumor cells subsequently restored their ability to suppress T cell activation.
Using RNAseq and qPCR, the researchers identified downregulation of genes encoding glycolytic pathways and glycolytic enzymes in Fto-Kd tumor cell lines. Investigating the transcription factors that control these genes, the researchers turned to the MYC and bZIP families, which are known to transcriptionally activate genes for glycolytic enzymes. While c-MYC was actually found to be upregulated, several bZIP family members, including c-Jun, JunB, and C/EBPβ were downregulated at both the RNA and protein levels. Forced overexpression of Junb in Fto-Kd tumor cells was sufficient to restore their glycolytic function and their suppression of T cell activation. A chromatin analysis using ATACseq further showed that loci for predicted enhancers of glycolysis were less accessible in Fto-Kd cells. Together, these results suggested that knockdown of FTO repressed the expression of glycolytic transcripts.
Diving deeper into how FTO knockdown repressed glycolysis, the team looked at m6A methylation, which is closely related to the decay of m6A-marked RNA transcripts. When they mapped the m6A methylomes of B16-OVA cells using m6Aseq, they found that Fto-Kd cells showed a moderate increase in overall m6A methylation peak number, which was more pronounced around stop codons. Further, mRNA for bZIP family members was highly marked by m6A peaks, suggesting accelerated degradation of these transcripts. Active FTO was required to prevent the accumulation of m6A on these transcripts, suggesting that FTO promotes the expression of bZIP family transcription factors by demethylating these transcripts, thus delaying their degradation. Further supporting this, silencing Ythdf2, which has been suggested to play a role in the decay of m6A-marked genes, increased the expression of c-Jun, JunB, and C/EBPβ in Fto-Kd cells.
While FTO inhibitors have demonstrated antitumor effects in prior studies, their ability to enhance T cell activation has not been investigated. Building on their previous research, the researchers developed an optimized FTO inhibitor, Dac51, that was more potent than previous FTO inhibitors and showed enhanced binding to the FTO protein. When tumor cell lines were treated with Dac51, they showed an accumulation of m6A, increased degradation of m6A-marked transcripts, and reduced expression of c-Jun, JunB, and C/EBPβ at both the RNA and protein levels.
In vitro, administration of Dac51 to cocultures of tumor cells and T cells enhanced T cells’ production of cytokines and increased their cytotoxic killing capacity. To confirm activity in human tumors, patient-derived organoids were utilized. In this setting, Dac51 decreased expression of c-Jun, junB, and C/EBPβ, along with other genes encoding glycolytic enzymes in tumor cells, and enhanced activation in T cells.
In immunocompetent (but not immunodeficient) mice bearing B16-OVA tumors, Dac51 increased the proportion of tumor-infiltrating CD8+ T cells, increased IFNγ production, and inhibited tumor growth to an extent similar to Fto-Kd. In mice bearing MC38 tumors, Dac51 slowed tumor growth and prolonged survival. Combination with anti-PD-L1 further enhanced survival and induced complete regressions in some mice. When surviving mice were rechallenged using 10 times more tumor cells than the initial inoculation dose, all mice showed complete regressions, suggestive of long-term memory.
Together these results suggest a mechanism by which FTO mediates demethylation of mRNA transcripts in tumor cells, which delays degradation of RNA encoding bZIP family genes and increases expression of these transcription factors. These transcription factors then go on to promote the expression of genes involved in glycolysis in tumor cells, contributing to an immunosuppressive tumor microenvironment that impairs T cell activation. This mechanism can be disrupted by targeting FTO, thereby increasing m6A methylation, reducing tumor cell glycolysis, and enhancing T cell activation and antitumor immunity. This strategy can further synergize with PD-L1 checkpoint blockade and could potentially be used to enhance immunotherapy in patients.
by Lauren Hitchings



