Constitutive signaling from a single mutation in the KIT proto-oncogene can be sufficient to drive the formation of a gastrointestinal stromal tumor (GIST). These tumors constitutively express immunosuppressive IDO, which dampens dendritic cell-primed effector CD8+ T cell responses. Previous studies have suggested that using imatinib, a small molecule Kit inhibitor that dampens IDO expression, alone or in combination with PD-1/PD-L1 checkpoint blockade could be effective in treating GIST. In practice, however, imatinib falls short. Despite signs of enhanced T cell activation in the early stages of imatinib therapy, combination with checkpoint therapy showed only very limited benefit, suggesting induced defects in T cell-mediated immunosurveillance and tumor control. In a study recently published in the Journal of Experimental Medicine, Medina et al. explored the mechanisms behind why Kit inhibition fails as an immunotherapy with chronic use, and investigated strategies that may help to redeem the seemingly promising effects of imatinib on antitumor immunity.
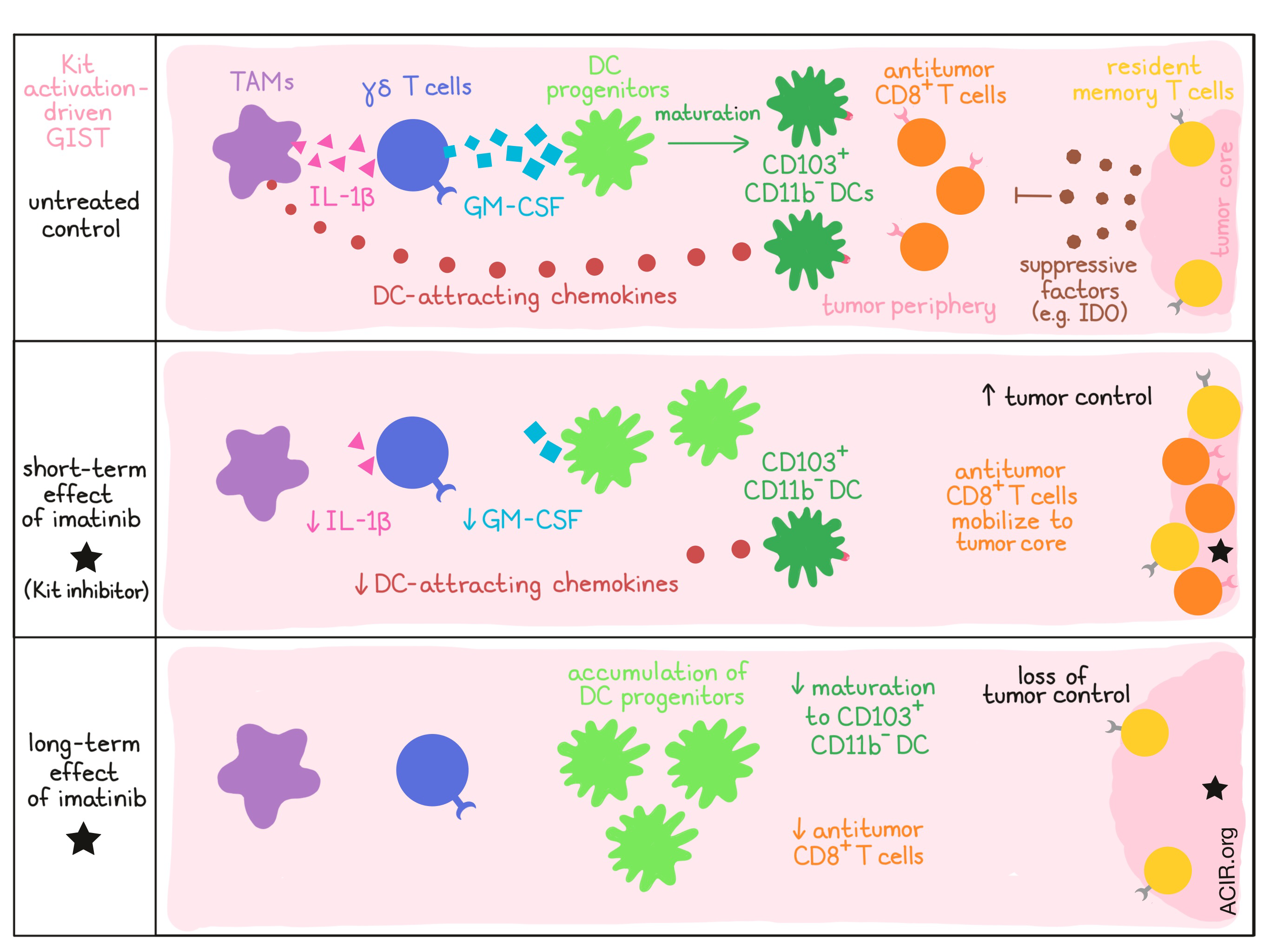
First, Medina et al. created a mouse model of Kit activation-induced GIST that closely mimicked human GIST. Using this model, the researchers explored the dendritic cell compartment and found that within tumors, tissue-resident/migratory CD103+CD11b- DCs (analogous to CD141+ DCs in humans) were the most frequent DC subset, that they were mature (expressing CD80, CD86, and CD40), and that they were distributed heterogeneously, often clustering in the vicinity of vascular structures along the tumor periphery. In Batf3-/- knockout mice, inherent tumor control was reduced, and treating the mice with imatinib induced no further effect, indicating that tumor control and the antitumor effect of imatinib are partially dependent on CD103+CD11b- DCs.
Looking next at the CD8+ T cell compartment in GIST-bearing mice, the researchers observed heterogenous distribution patterns similar to those of DCs: activated CD8+ T cells clustered in the periphery and around vascular structures, though additional clusters – later identified as resident memory T cells – were observed surrounding the tumor core. In Batf3-/- mice, CD8+ T cells in the periphery were reduced, but the population of resident T cells at the core remained unaffected; depletion of the remaining CD8+ T cells had no further effect on tumor growth, confirming that CD8+ T cells primed by Batf3-dependent DCs were responsible for tumor control.
Looking closer at what happens following treatment of GIST-bearing mice with imatinib, the researchers observed a dramatic reduction of CD103+CD11b- DCs in mice after just one week of treatment. Imatinib also reduced CD8+ T cells in perivascular regions at the tumor periphery, and increased CD8+ T cells at the core, where the resident subtype was no longer dominant, indicating that imatinib had mobilized antitumor CD8+ T cells. By week 4, however, CD8+ T cells had reduced in both the periphery and the tumor core, with remaining T cells lacking markers of maturity and antigen experience. The researchers therefore concluded that early on, imatinib had mobilized a pre-existing immune response, but that its negative effect on CD103+CD11b- DCs rendered mice unable to maintain a long-term antitumor response.
Looking at the effect of imatinib on the broader DC lineage, Medina et al. found that the reduction of CD103+CD11b- DCs following imatinib treatment was an on-target effect directly related to Kit inhibition in the tumor microenvironment. The researchers then identified an accumulation of CD24hiSIRPα- DC progenitors, which had become the dominant DC population in tumors by week 4. Hypothesizing that imatinib cause defective DC maturation, the researchers looked at GM-CSF, a growth factor that controls the differentiation of cells of the myeloid lineage, and found it was depleted in the tumor following a week of imatinib treatment. Antibody-mediated depletion of GM-CSF in control mice had similar results to imatinib treatment, and there was no additive effect of imatinib and GM-CSF depletion. Together, this indicated that imatinib halted the differentiation of Batf3-lineage DC progenitors into CD103+CD11b- DCs by depleting GM-CSF.
Working backwards through the mechanism, Medina et al. found that GM-CSF was produced almost exclusively by γδ T cells and that depletion of γδ T cells had similar effects to GM-CSF depletion or treatment with imatinib. Imatinib treatment did not, however, affect the number of γδ T cells in the tumor, so the researchers concluded that imatinib must instead be affecting γδ T cell function. They then found that IL-1β, produced by tumor-associated macrophages (TAMs), controlled the expression of GM-CSF by γδ T cells, and that depletion of IL-1β had effects similar to imatinib treatment, GM-CSF depletion, or γδ T cell depletion. Overall this indicates that imatinib reduces macrophage production of IL-1β, which reduces γδ T cell production of GM-CSF. As GM-CSF is required for DC maturation, this causes an accumulation of DC progenitors and a reduction of mature CD103+CD11b- DCs, which leads to a reduction of antitumor CD8+ T cell responses and a loss of antitumor efficacy.
While the depletion of IL-1β or GM-CSF had effects similar to that of imatinib, the effect of imatinib on the decrease in CD103+CD11b- DCs was still stronger, suggesting that imatinib also functioned through an additional mechanism. Exploring alternative avenues, the researchers found that imatinib treatment also led to reduced TAM expression of chemokines associated with DC recruitment (including CXCL9, CXCL10, CCL3, and CCL4) within tumors, possibly accounting for the remaining impact of imatinib treatment.
Because imatinib has shown some antitumor efficacy alone and in combination with checkpoint blockade, the researchers explored possible strategies to redeem this treatment. They found that expansion of DC progenitors from the bone marrow using FLT3L and the addition of poly I:C (to induce DC maturation) to imatinib led to increased expression of costimulatory markers on CD103+CD11b- DCs and their progenitors, increased activation of effector CD8+ T cells, and improved antitumor immunity.
by Lauren Hitchings



