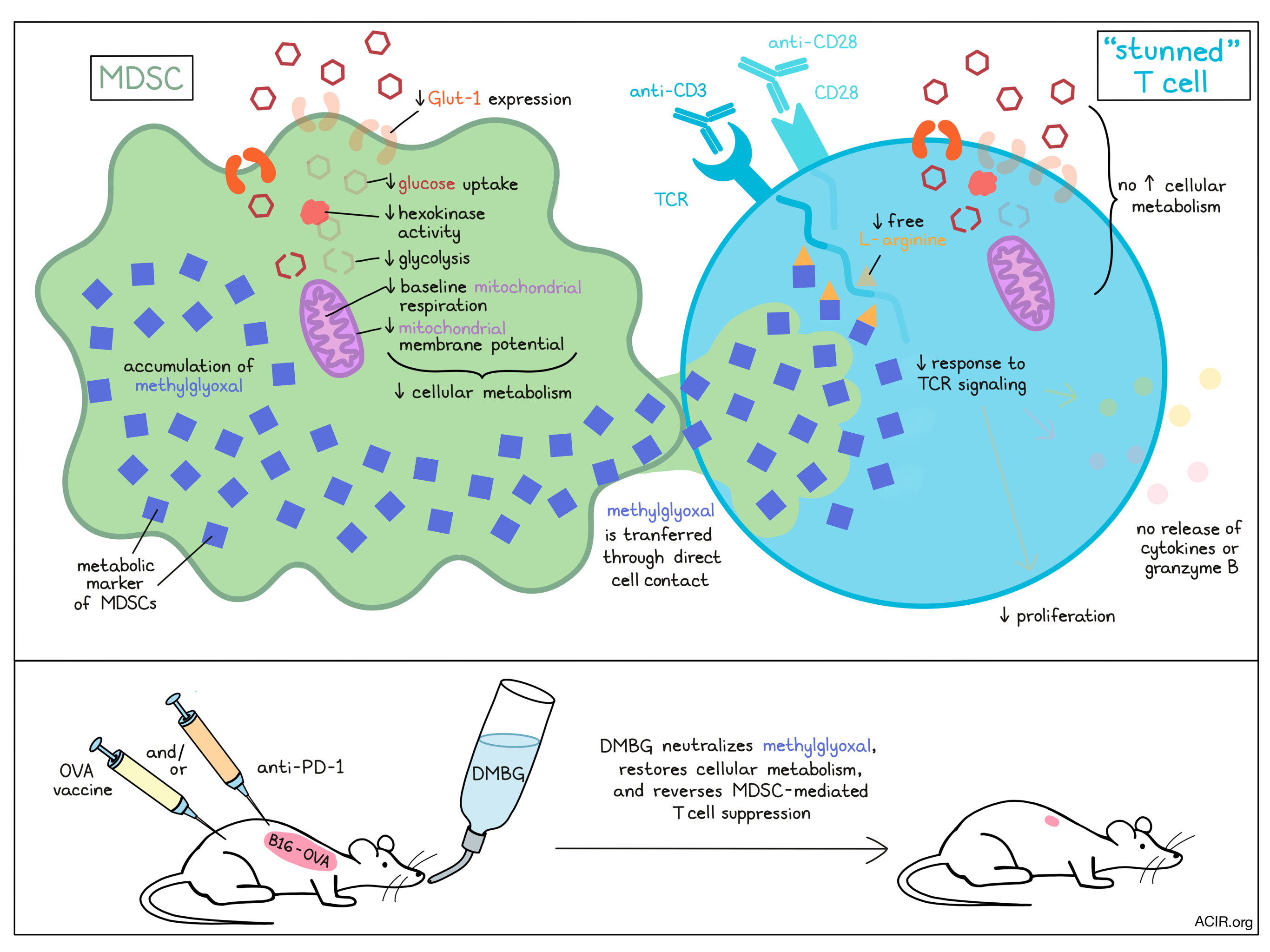
Suppressive myeloid cells, namely myeloid-derived suppressor cells (MDSCs), are known to appear in situations of chronic inflammation and cancer, where they suppress cytotoxic T cell responses. However, without an existing definitive marker for this immune subset, MDSCs can be difficult to study and target. In research recently published in Nature Immunology, Baumann et al. not only present evidence of a metabolic marker for MDSCs, but also show how that marker plays a role in the suppression of effector T cell functions.
To create a model of MDSCs to study, Baumann et al. used culturing with a human stromal fibroblast cell line to convert human peripheral blood monocytes into MDSCs that were phenotypically similar to those found in cancer. Transcriptome analysis showed 200 differentially expressed genes between the monocytes and MDSCs, and while it did not reveal a singular marker that was sufficient to differentiate the two cell populations, it did reveal downregulation of genes encoding glycolysis-related enzymes in MDSCs. In vitro, model MDSCs showed reduced glucose uptake, reduced surface expression of Glut-1 (the transporter that mediates glucose uptake in immune cells), lower hexokinase activity, an inability to utilize glucose for glycolysis, reduced mitochondrial membrane potential, and lower baseline mitochondrial respiration. Reduced glucose uptake and hexokinase activity were confirmed in human tumor-derived MDSCs. Together this showed that cellular metabolism is strongly reduced in MDSCs.
Looking into how in vitro-generated MDSCs interact with T cells, the researchers cocultured the two cell types and found that following interactions with MDSCs, T cells had reduced phosphorylation of key protein kinases downstream of TCR activation, suggesting suppression of TCR signaling. TCR and CD28 signaling typically increase glucose uptake and glycolysis within T cells, however, when T cells were stimulated with anti-CD3 and anti-CD28 in the presence of MDSCs, they failed to increase Glut-1 surface expression, glucose uptake, hexokinase activity, glycolysis, or mitochondrial respiration. As a result, ATP concentrations were reduced, and the T cells were unable to proliferate, express cytokines (including IFNγ and TNF), or release granzyme B. Tumor-derived MDSCs, but not monocytes, had the same suppressive effect, essentially rendering T cells unable to respond to activation.
Next, Baumann et al. observed that MDSCs were not able to suppress T cell activation when the two cell types were separated by a transwell, suggesting that the suppressive activity depends on direct cellular contact. Further investigating this interaction, they used dyes to label various cellular components of MDSCs and found that cytosolic constituents (but not entire organelles) from MDSCs transferred into nearby CD8+ T cells, as well as CD4+ T cells and NK cells, though no transfer of surface molecules was observed. Similar results were observed in tumor-derived MDSCs and an in vivo tumor model.
Investigating how the interaction between MDSCs and T cells halts T cell activation, Baumann et al. found that dimethylbiguanide (DMBG) reversed the MDSC-mediated suppression of T cell proliferation and reinstalled T cell signaling. Based on knowledge that guanidine neutralizes dicarbonyls, the researchers then searched for this class of metabolites within MDSCs and identified an accumulation of methylglyoxal in both model and patient-derived MDSCs. The accumulation of methylglyoxal was found to be due to semicarbazide-sensitive amine oxidase (SSAO) acting on aminoacetone. Importantly, the accumulation of methylglyoxal was highly specific to MDSCs and was not found in other human immune cell populations, suggesting that it could be used as a clear molecular identifier for MDSCs. When model MDSCs were incubated with DMBG, the accumulation of methylglyoxal was lost, the capacity to take up glucose was regained, and glycolysis and mitochondrial respiration were restored, suggesting that methylglyoxal also plays an important functional role in MDSC metabolism.
Using a methylglyoxal-sensitive fluorescent reporter to investigate whether methylglyoxal was involved in T cell suppression by MDSCs, the researchers found that methylglyoxal could be detected in CD8+ T cells within ten minutes of being in contact with MDSCs. When MDSCs were pre-treated with DMBG, however, methylglyoxal was not detected in T cells, and T cells remained capable of taking up glucose. Further, pretreating either MDSCs or CD8+ T cells with DMBG prevented the suppression of activation-induced cytokine production and granzyme B release in CD8+ T cells, suggesting that DMBG abrogated metabolic suppression in both cell types. DMBG treatment also shortened the amount of time it took for T cells suppressed by MDSCs to regain their capacity to respond to activation.
Methylglyoxal is known to react with the amino acid L-arginine, which is required for T cell activation. Investigating whether this interaction might play a role in the suppression of T cell activation, the researchers looked at T cells following the transfer of methylglyoxal from MDSCs, and found that L-arginine was completely reduced and that methylglyoxal-derived glycation products of L-arginine were increased, suggesting that methylglyoxal depletes L-arginine and may render L-arginine-containing proteins non-functional.
Looking at samples from patients with liver cancer, Baumann et al. found that MDSCs from tumors were stronger at suppressing CD8+ T cell proliferation than MDSCs from peritumoral tissue or blood. DMBG reversed the suppressive effect of all human M-MDSCs. PMN-MDSCs could not be sufficiently isolated for testing, however, those from blood did not show evidence of methylglyoxal accumulation or suppressive activity.
Finally, to determine whether the mechanism they had uncovered could be utilized to enhance immunotherapy, Baumann et al. tested a therapeutic OVA vaccine, DMBG treatment, and anti-PD-1 in mice with B16-OVA melanoma. None of the treatments were effective as monotherapies, however, the combination of the OVA vaccine with DMBG treatment reduced tumor growth, and the combination of DMBG treatment with anti-PD-1 strongly reduced tumor growth. Characterization of the tumor immune contexture showed that most of the MDSC-like cells showed evidence of methylglyoxal accumulation, displayed an inability to take up glucose, and suppressed CD8+ T cell proliferation when cultured ex vivo. In DMBG-treated mice, these effects were not observed.
Overall, Baumann et al. report that methylglyoxal is a metabolic marker that accumulates in MDSCs and greatly reduces metabolic functions. They also uncovered that MDSCs transfer methylglyoxal to T cells through direct cell contact, rendering T cells metabolically dysfunctional and temporarily “stunned” – unable to respond to T cell activation signals. This occurs because methylglyoxal depletes L-arginine and may affect the production of L-arginine-derived protein products. Neutralization of methylglyoxal using DMBG overcame MDSC-mediated T cell suppression and, in a murine tumor model, improved the efficacy of immunotherapies.
by Lauren Hitchings
Meet the researcher
This week, lead author Bastian Höchst answered our questions.

What prompted you to tackle this research question?
The basic idea was that I always had problems with the idea that MDSCs are suppressive via NO, ROS and arginase, especially since other cells (type I macrophages, granulocytes, hepatocytes) produce significantly more of these substances/enzymes and are not generally suppressive. Therefore, I assumed that there must be one or more mechanisms besides these factors.
What was the most surprising finding of this study for you?
The biggest surprise was the fact that there is a cytosolic exchange of substances, which also happens very quickly. We observed that this transport could happen within minutes, and that this exchange seemed to take place relatively often, at least between immune cells, with sometimes massive effects.
What was the coolest thing you’ve learned (about) recently outside of work?
The most interesting thing I have learned outside of work is that genes have a great influence on the behaviour and nature of children. Apparently, even behavioral traits are inherited in this way.



