
The ACIR team attended the 22nd CIMT Annual Meeting 2025 in Mainz, Germany. This week’s extensive special feature covers select talks from the conference. We have organized the content by topics below.
CIMT Lifetime achievement award
Rolf Kiessling
Keynote address
Cornelis JM Melief
Immune response to cancer
Barbara Maier
Karina Silina
Matthew Spitzer
Ronald Germain
T cell response and cellular therapies
Susanna Minguet
Lukas Bunse
Sine Reker Hadrup
Guangyuan Li
Cytokine therapies
Dario Neri
Ayline Kübler
Cancer antigens
Nikolaos G. Sgourakis
Angela Mauriello
Bispecific antibodies
Jan-Jaap Verhoef
Maren Köhne
CIMT Lifetime achievement award
CIMT Lifetime achievement award - Rolf Kiessling, Karolinska Institutet/Karolinska University Hospital
The recipient of the CIMT Lifetime Achievement Award for 2025 was Rolf Kiessling for his work in 1975, opening up the field of NK cells as important players in cellular immunology. Rolf Kiessling was graciously introduced by Michael Nishimura. Beyond the important science, which Nishimura was proud to have collaborated with him on, Nishimura also presented a bit of the personal side of Kiessling, including his eclectic interests, from fishing, sailing, and outdoor activities, to being a drone enthusiast and having pet alligators! Kiessling began by describing his early work in the laboratory of Eva Klein, a pioneer in tumor immunology and a mentor he had great respect for, at the well respected Department of Tumor Biology at the Karolinska Institutet in Stockholm. His work began at a time when T cells were gaining momentum, but before concepts like the T cell antigens and the T cell receptor were understood. In trying to understand the origin of the specificity of tumor rejection by prior tumor cell line immunization, the fortunate choice of the YAC-1 murine lymphoma cell line (low in MHC-I expression, as was later appreciated) to work with, and the unexpected ("that's funny" as a marker of discovery – Isaac Asimov) ability of spleen cells from non-immunized control mice to kill this cell line led to the discovery of “a new, previously undefined “cytotoxic cell”, aptly named Natural Killer (NK) cells. The NK cell field grew slowly, but was accelerated by the work of Klas Kärre, the first student of Kiessling, who outlined the bold “missing self” hypothesis as the basis of NK cell recognition and killing. An important detour in Kiessling's career was his work at the Armauer Hansen Bioresearch Institute in Ethiopia, Africa on leprosy, which provided the opportunity to conduct a clinical trial with IL-2 in treatment of leprosy. As the “golden age” of immunotherapy was ushered in with the discovery of checkpoint inhibition, Kiessling continued to have an impact in the field, working on how tumors evade and escape from immune control, and the immunosuppressive roles of MDSCs and ROS, and discovering approaches to counter these effects (e.g., with Nrf2-activating compounds). Finally, Kiessling continues to be active in the development of TIL therapies, and follows the advances in NK cell therapy during the now “golden age of cell therapy of cancer”
Keynote Address
Keynote lecture: Conditions for successful therapeutic vaccination of late stage cancer - Cornelis JM Melief, Emeritus Professor, Leiden University, The Netherlands
In his Keynote Lecture, Cornelis (Kees) Melief, a long term-giant in the field of cancer vaccines, presented an overview of efforts, with the objective of proving that cancer vaccines are here to stay – at least in one case aided by a biomarker – despite the rollercoaster of excitement and lack of excitement over the years. Multiple antigens have been used to develop cancer vaccines, and the emergence of genomics technology has shown that cancer antigens arising from mutations, particularly in cancer types where the mutation burden is high, such as sunlight-driven melanoma, are an exciting new area. Melief focused on viral-driven cancers, which represent about 10-15% of worldwide cancers. Among the various types of immunotherapy, vaccines are unique in their ability to create long-lasting immune memory, and don't require genetic manipulations, which should simplify preparation. A variety of cancer vaccine platforms and delivery routes have been and are currently being tested, but the fundamental requirements are delivering high concentrations of antigen to dendritic cells, which in the lymph nodes, stimulate strong and sustained Th1 T cells and CTLs capable of migrating to and acting in the tumor. Cooperation and communication between CD8+ T cells, CD4+ T cells, and dendritic cells is required for optimal effector function and memory. Melief then described three highly encouraging recent studies with exciting clinical outcomes: two personalized neoepitope mRNA vaccines in melanoma and pancreatic cancer, and a study with synthetic long-peptide vaccines in renal cell cancer, all in the adjuvant setting, which is emerging as an ideal setting for therapeutic cancer vaccines. Synthetic long peptides have multiple advantages compared to other forms of antigen, including bypassing transcriptional and/or translational hurdles and requirements for processing (which can only occur in professional antigen processing cells), and exhibit clearly better immunogenicity than comparable full-length proteins. Melief then presented the results of a trial with IB101, synthetic long peptides targeting the E6 and E7 oncogenes of human papilloma virus (HPV). Early encouraging results in pre-malignant, HPV-induced grade 3 vulvar intraepithelial neoplasia led to a study in late-stage HPV+ cervical cancer, where standard-of-care chemotherapy lowered circulating suppressive myeloid cells, leading to strong and durable vaccine-induced T cells. Immune responses above the median were associated with improved overall survival. A phase II study in HPV+ oropharyngeal carcinoma (OPC) of IB101 with nivolumab showed a response rate twice that of nivolumab alone, leading to a set of studies (one of which was randomized, double-blind, and placebo-controlled) of IB101 with cemiplimab in HPV+ OPC and cervical cancer. Although there was no significant change in overall response rate (ORR), a pre-treatment combined PD-L1 Score (CPS) of ≥20 emerged as a marker identifying patients with statistically significantly improved ORRs and overall survival. Poor ECOG performance status, often associated with neutrophilia, was a marker of poor response. Melief postulated that vaccine-induced, target-specific T cells produced IFNγ, which upregulated PD-L1, making the tumor cells more sensitive to anti-PD-1 therapy. This notion was backed by findings in various patient cohorts treated with precursors of IB10. Encouragingly, in patients with HPV+ head and neck cancer who had progressed on prior anti-PD-1 therapy, the combination of the E6/E7 vaccine and cemiplimab resulted in significant long-term stable disease. To wrap up, Melief concluded that therapeutic cancer vaccines are a safe and optimal immunotherapy platform with the ability to target intracellular proteins. Vaccines may be most active when their targets are oncogenes critical to tumor cell survival, in the setting of minimal residual disease, in combination with immune checkpoint blockade, and when proper biomarkers are present, one of which is inflamed tumor tissue.
Immune response to cancer
Do infiltrating monocytes regulate anti-tumor T cell immunity in tumor-draining lymph nodes?- Barbara Maier, The Research Center for Molecular Medicine of the Austrian Academy of Sciences, Austria
Babara Maier and her research team explored how tumor progression and metabolic and cytokine signals within the tumor interstitial fluid, traveling via lymphatic vessels to lymph nodes, remodel the spatial niches in tumor-draining lymph nodes (tdLNs) to enhance local metastasis. Using an orthotopic oral carcinoma model and imaging mass cytometry with 26 markers, Maier and her team compared whole tissue cross-sections of naive cervical lymph nodes with early and advanced tdLNs, enabling linkage of LN architecture with detailed cellular features. PCA analysis revealed that as tumors developed, myeloid cells infiltrated the tdLNs, the T cell zone shrank and fragmented, with a reduction in T cells, and the distance between myeloid cell clusters and T cells decreased. scRNAseq of the myeloid compartment demonstrated a marked increase in IL-1bhi myeloid cells in both early and advanced tdLNs. These IL-1bhi myeloid cells were primarily monocytic in origin (although some neutrophil markers were present) and expressed gene signatures of immaturity, immunosuppression, and inflammation (a myeloid suppressive signature). A similar IL-1bhi myeloid cell cluster was observed in tdLNs obtained from patients with oral carcinoma. Manual histologic annotation of lymph node domains revealed a substantial expansion of the lymphatic domain in advanced tdLNs. Infiltrating monocytes accumulated in this lymphatic domain, without migrating into the T cell zone. Concurrently, dendritic cells and stromal cells increased in the T cell zones, and interactions between various T cell subsets, dendritic cells, and activated fibroblastic reticular cells* (FRCs) were strongly upregulated, suggesting an ongoing immune response, despite the shrinkage and fragmentation of the T cell zone. Cellular triplet analysis unveiled an interaction between the monocyte/macrophages cluster, the lymphatic endothelium, and proliferating Ki67+CD8+ T cells within the lymphatic domain. Unbiased neighborhood analysis confirmed the expansion of the lymphatic niche with infiltrating myeloid cells, and the compression of the deep T cell zone. While proliferating Ki67+ T cells were primarily located in the deep T cell zones of naive lymph nodes, they were also found in the lymphatic domain and, in particular, in the blood vascular niche (located between the deep T cell zone and the lymphatic/myeloid niche) in early and advanced tdLNs. During tumor progression, the numbers of dendritic cells and activated FRCs* increased in the T cell zone, but significantly decreased in the lymphatic/myeloid niche. This suggests a depletion of these immunostimulatory, antigen-presenting cells as the putatively suppressive monocytes infiltrated this space. DCs and activated FRC remained highly abundant in the blood vascular niche throughout tumor progression. These interesting findings left Maier and her team with many important follow-up questions that will now be studied in her lab.
Survival-relevant features of the immune microenvironment in lung cancer - Karīna Silina, ETH Zurich
Karīna Silina described her deeper investigation into the development, function, and clinical relevance of tertiary lymphoid structures (TLSs) in tumors. As recently observed, the dogma of the unidirectional flow of T cells from induction in lymph nodes to action in tumors is now challenged by the possibility that TLSs circumvent that flow and induce T cells directly in the tumor. The development of these organized multi-cell structures, which form during chronic inflammation and within the tumors, mimic lymphoid neogenesis and provide access to antigenic stimulation and priming that can support effector cells and the release of antibodies targeting the cancer. Earlier work with lung tumors demonstrated that TLS development is similar to that observed in lymph node development, proceeding in a stepwise manner, initiating from dense, unorganized aggregates of T cells, follicular DC cells, and stromal cells. The presence of such structures in the tumor microenvironment (TME) correlates with increased survival of patients, and studies in mice demonstrate an important role in pathogen clearance, which can occur even in lymph node-deficient mice. Silina has attempted to understand how these important TLSs develop, why they do not develop in some tumors, and, why some patients with such structures, do not benefit. Taking a GeoMx spatial transcriptomics approach allowed correlation of gene pathways enriched in early and mature TLSs. This revealed that mature TLSs were characterized by enrichment in signatures for cytoplasmic translation (a sign of strong T cell activation), as well as T cell cytotoxicity, and these two pathways were highly enriched in patients demonstrating long-term survival. A 25-plex CODEX panel was then used to confirm these findings at the single-cell level and to compare TLS-high patients exhibiting short- or long-term survival. Long-term survivors were characterized by high PD-L1 expression in areas surrounding tumor nests (“PD-L1 cuffs”). These “cuffs” were almost exclusive to long-term-survival patients, and dimensionality reduction indicated that these “cuffs” serve as sites of CD8+ T cell, plasma cell, and dendritic cell interactions. Stem-like T cells (PD-1+ and TCF1+) appeared to reside mostly in the T cell cluster-rich immune niches, particularly where B cells are also infiltrating, consistent with other data indicating the need for immune cell niches for the survival of stem-like cells. Long-term survivors were particularly enriched in TLSs with switched memory B cells and proliferating and activated CD8+ T cells. In long-term survivors, TOX expression was high across T cells in multiple regions of the tumor, consistent with other data that TOX expression is not solely related to exhaustion. In short-term survivors, the frequencies of TOX+ cells and switched memory B cells were reduced. Overall, a more refined understanding of TLSs may provide novel prognostic markers and identify approaches to induce such effective structures.
Systemic and local immune responses to cancer immunotherapy - Matthew Spitzer, UC San Francisco, USA
Matthew Spitzer asked the question of where T cells are activated following immunotherapy. Early research from Spitzer's lab suggested that immune cell proliferation following immune checkpoint blockade (ICB) primarily occurs in the lymph nodes, and is not sustained within the TME, based on experiments blocking immune cell egress from the lymph node (LN) with FTY720 during ICB, which diminished treatment efficacy in MC38 tumor models. ICB is known to primarily reactivate progenitor exhausted cells (Tpex), while terminally exhausted cells (Tex) cannot be reactivated due to epigenetic modifications. CYTOF analysis of paired tumor and LN samples from patients with HNSCC revealed an enrichment of two Tpex-like clusters co-expressing TCF1 and PD-1 in tumor-draining LNs (tdLNs) compared to primary tumors. Further scRNAseq coupled with T cell receptor sequencing demonstrated clonal relationships between T cells in the LNs and tumors. Notably, T cells with shared TCRs infiltrating the TME displayed a more pronounced terminally exhausted gene signature than found in the LN, suggesting that T cells in the LNs retain a more progenitor-like state and replenish the T cells in the TME, which become exhausted. To assess the impact of ICB, a neoadjuvant clinical trial was conducted in which 9 patients with HNSCC received anti-PD-L1 therapy prior to surgery. Multiplexed spatial imaging of uninvolved LN tissues revealed a decrease in the relative abundance of Tpex cells in treated patients, accompanied by a trend towards an increase in intermediate exhausted cells (Tex-int). Tex-int cells were more frequently located in proximity to Tpex cells after treatment, suggesting local differentiation within the LN. Moreover, Tpex and Tex-int exhibited increased PD-1 expression, indicative of active T cell receptor signaling. DCs were more commonly found in the vicinity of Tpex cells after treatment, suggesting a potential role for DCs in Tpex activation. In contrast, baseline analysis of metastatic LNs showed a reduced frequency of Tpex and an increased abundance of Tex, resembling the TME. Following anti-PD-L1 treatment, clustering of Tpex and Tex-int cells, and upregulation of PD-1 were not observed in metastatic LNs. DCs surrounding Tpex in metastatic LNs upregulated various inhibitory molecules, adopting a more tolerogenic phenotype, and Tregs surrounding Tpex expressed high levels of Foxp3 and Tim3, further contributing to an immunosuppressive environment. These findings suggest that while regional LNs support Tpex cell maintenance and early response to ICB; this process is impaired in metastatic LNs. To identify opportunities for productive manipulation, mining public data revealed differential expression of the IL-7Rα (CD127) between Tpex and Tex. Consistent with this, analysis of patient samples showed significantly higher CD127 expression on Tpex cells. Preclinical studies using a long-acting IL-7 variant (NT-I7) in oral SCC models demonstrated significant increases in LN size, CD8+ T cell counts, and T cell proliferation in tdLNs, with a subsequent influx of CD8+ T cells into the TME. Importantly, NT-I7 did not impact Treg levels in the TME. This treatment led to tumor regression in an immunogenic model, and delayed tumor growth in a less immunogenic model. CYTOF analysis revealed that Tpex cells exhibited the strongest proliferative response to NT-I7, differentiating into a distinct population characterized by high expression of granzymes A and B, termed exhausted effector-like cells. Tex cells were reduced following NT-I7 treatment, and the effector-like cells regained the ability to produce IFNγ and TNFɑ, as well as exhibit enhanced in vitro tumor cell killing. Combination therapy with ICB and NT-I7 showed additive benefits. In a small neoadjuvant trial in three patients with recurrent or metastatic HNSCC, a single dose of NT-I7 prior to surgery significantly expanded CD8+ T cells within the TME, along with a trend towards increased Tpex, a significant increase in effector-like cells expressing granzymes A and B, and a trend towards reduced Tex. These findings suggest that IL-7 receptor agonism can promote a more effective antitumor immune response by preferentially activating Tpex cells and driving their differentiation towards a functional effector-like state, potentially synergizing with ICB.
Using highly multiplex 2D and 3D optical imaging for spatially-resolved analysis of the immune response to cancer - Ronald Germain, National Institute of Allergy and Infectious Diseases National Institutes of Health, USA
With an underlying recognition that normal tissue function and immune reactions occur in complex tissues, Ronald Germain emphasized the necessity of employing high-information-content, spatial cell organization analysis through both dynamic and static multiplexed imaging techniques. A routine 30–50-plex imaging panel was developed that could probe both surface and intracellular targets at the level of cytoplasmic and nuclear localization of phosphorylated proteins, and even monitor RNA. These panels enabled the field of “Histo-Cytometry”, in which quantitative, and now spatial data could be developed for individual cell types at the cell signaling state, phenotypic, and functional levels. Comparing Histo-Cytometry to Flow Cytometry (representative of methods that rely on single cells), similar levels of CD4+ and CD8+ lymphocytes were detected, but there were very significant increases in the detection of dendritic cells and doublets using Histo-Cytometry. Applying this approach to analysis of a “hot”, immunotherapy-sensitive, transplantable pancreatic cancer cell line developed from a GEMM tumor, Germain showed that without therapy, these tumors exhibited high Treg levels. Two days after treatment with anti-PD-1, anti-CTLA-4, and an agonist anti-CD40 antibody, Tregs were mostly cleared from the tumors, and the tumors subsequently resolved. Agonist anti-CD40 binding to cDC1s, but not anti-CTLA-4, was essential for this effect. Foxp3 lineage tracing showed that therapy did not induce Treg death, but a loss of Foxp3 expression in those cells and conversion into ex-Tregs. This effect was restricted to the tumor microenvironment, and was dependent on IL-12 and IFNγ. The ex-Tregs became Tbet+ effector Th1 cells that produced IFNγ and were in close contact to cDC1s. High-resolution imaging of NFAT showed that ex-Tregs had the highest proportion of cells with cytoplasmic-to-nuclear NFAT translocation (20x increase), a marker of TCR signaling. Therefore, the Tregs that were being converted with the therapy were among those with the greatest functional TCR engagement, and would be highly suppressive if not converted. Thus, agonist anti-CD40 acting on cDC1s was rebalancing the immunosuppressive TME. Similar nuclear NFAT translocation could also be identified in human samples in vitro. To more deeply probe tissues, Germain described work with high-plex imaging by confocal microscopy using an open-source technique called iterative bleaching extends multiplexity (IBEX), which utilizes readily available reagents. Up to 8 parameters (up to 12-15 with special equipment) can be used per cycle, currently allowing up to 82 parameters to be analyzed. To make sense of the data, the researchers developed an algorithm working at the pixel level and using machine learning to identify individual cell phenotypes. To analyze cellular interactions and signal propagation across different scales, from local interactions to gradients spanning large tissue regions, the software tool spatial analysis of cell ensembles (SPACE) was developed, allowing robust evaluation of 2, 3, or 4 cell clusters, as well as gradients within tissues. As an example, Germain described an analysis of CD4+ T cell inhibition of tumor cell growth using SmartA cells, which are specific for a CD4 epitope of the CMV glycoprotein GP, and recognize MC38-GP cells. High-resolution imaging demonstrated that SmartA cells killed tumors not directly, but by producing IL-3, which enhanced macrophage production of TNF, which acted on the host vasculature, causing vascular leak and tumor necrosis. Combining IBEX with a clearing method (Ce3D) allowed determination and visualization of cell interactions, signals, and gradients across tissues in 3D. Ce3D imaging of murine tumor and tumor-draining lymph nodes (tdLN) following adoptive transfer of OT-I cells revealed stem-like memory CD8+ T cells (PD-1+, TCF1+) in association with XCR1+ cDC1s in the tdLN, but not the tumor. Interestingly, after a single dose of anti-PD-1, these high-affinity stem-like memory cells were lost in the tdLN, raising the possibility that the impact of anti-PD-1 blockade occurs only on the first 1-2 doses.
T cell response and cellular therapies
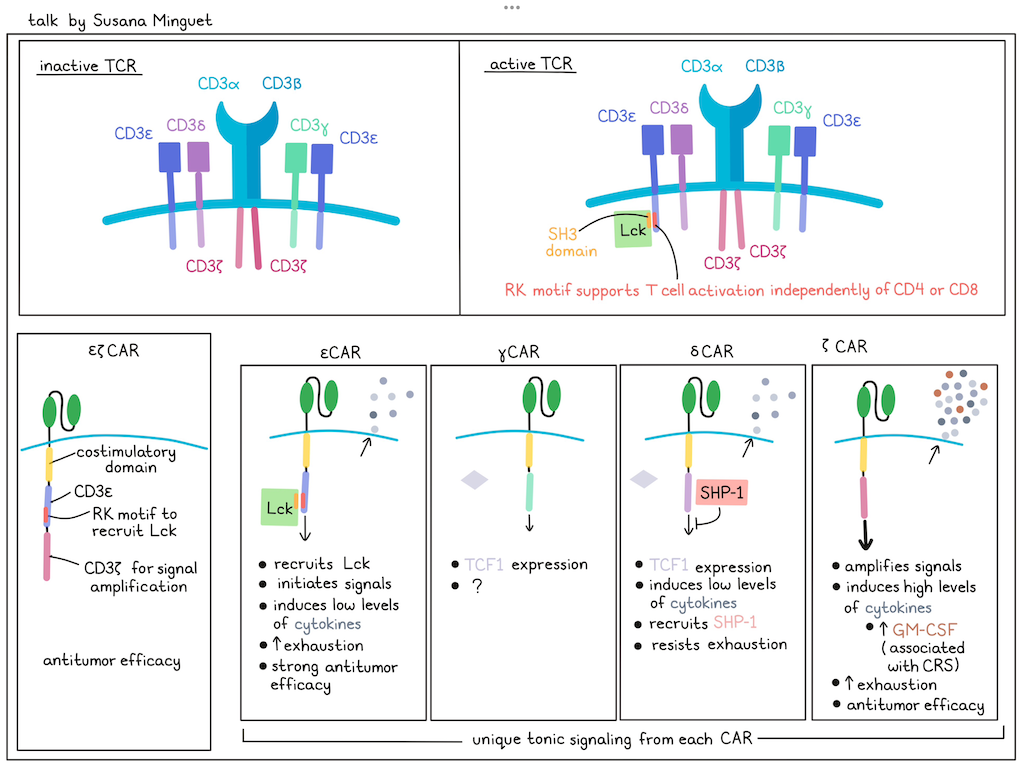
From TCR fundamental research to innovative immunotherapies - Susana Minguet, University of Freiburg, Germany
Although chimeric antigen receptors (CARs) are used successfully in the clinic, they still face limitations, such as inadequate CAR design or excessive activation that leads to toxicities, such as cytokine release syndrome (CRS). Current CARs using the CD3ζ chain fail to completely mimic T cell activation. To enhance CAR performance, Susana Minguet has turned to the structure of the native TCR signaling complex, hypothesizing that understanding the intricacies of TCR signaling could help to develop superior CAR candidates by selectively incorporating one or more of the diverse components. The TCR exists in (at least) two states: a predominant inactive resting state and an active signaling state, which requires phosphorylation mediated by the Lck kinase. While TCR activation typically requires co-stimulation via CD4 or CD8 to bring the Lck kinase into close proximity of the TCR complex, anti-CD3 antibodies can trigger the TCR independently of CD4/CD8 involvement. To investigate how anti-CD3 antibodies stimulate the TCR, Minguet and her team employed proximity ligation assays (PLAs) to visualize protein clustering. In resting T cells, minimal interaction between Lck and the TCR were observed. However, TCR activation via anti-CD3 significantly increased their proximity, confirming direct Lck recruitment to the TCR upon activation. Biochemical analyses revealed an interaction between the SH3 domain of Lck and the non-phosphorylated ε-chain of the TCR, and modeling indicated interactions at the arginine-rich receptor kinase (RK) motif of CD3ε. Mutation of these positively charged amino acids abolished Lck–TCR interactions and diminished downstream TCR signaling, confirming a novel mechanism for CD4/CD8-independent Lck recruitment to the TCR. To translate this to CAR design, Minguet introduced the Lck-interacting RK motif into a conventional CAR containing 4-1BB and CD3ζ signaling domains. In a xenograft “stress-test” mouse model (using reduced effector cell numbers), CARs incorporating the RK motif (RK-CARs) demonstrated improved tumor control and prolonged survival. Building upon this, Minguet explored the potential of utilizing the entire cytoplasmic domain of the different TCR signaling chains within CARs. Each CAR contained the extracellular domain and transmembrane region of a standard CAR, but differed in their intracellular signaling domain (ζ, ε, δ, or γ). All CAR constructs were expressed on the T cell surface and delivered tonic signals that were tail-specific, with very limited overlap in commonly regulated genes (ICOS-L and CCL22). While all TCR chain-based CARs exhibited comparable in vitro cytotoxicity, in vivo efficacy varied significantly. Surprisingly, CARs incorporating the δ-chain, and to a lesser extent the γ and ε chains, outperformed the conventional ζ-based CAR in preclinical tumor models. To understand these differences, IFNγ, TNF, IL-2, and GM-CSF secretion were measured upon target cell engagement. Consistently, ε- and δ-CAR T cells produced lower levels of cytokines compared to ζ-CAR T cells, which uniquely induced high levels of GM-CSF, a cytokine implicated in cytokine release syndrome, suggesting that ζ-based CARs may be more prone to inducing systemic toxicities. Further, δ-CAR T cells retained their antitumor capacity upon repeated antigen exposure, while ζ- and ε-CAR T cells exhibited signs of exhaustion. Mechanistically, δ-CAR T cells were found to maintain stable expression of SHP-1, a phosphatase that negatively regulates TCR signaling, suggesting that δ-signaling achieves a balance between activating and inhibitory signals, preventing premature exhaustion. Further, TCF1 was mainly expressed by δ- and γ-CAR T cells. Overall, each CD3 domain variant contributes different functionality to the TCR complex, extending the toolbox for CAR production.
Cellular Therapies for Primary Brain Tumors - Lukas Bunse, German Cancer Research Center DKFZ, Germany
Adult glioblastoma is a difficult-to-treat cancer characterized by a hostile tumor microenvironment, relatively few mutations, exclusion of tumor-infiltrating T cells, and no response to immune checkpoint blockade. Lukas Bunse started his talk by reviewing data from a patient with an initial benign pituitary adenoma who progressed to refractory disease and, as a last effort, was treated with pembrolizumab, which durably reduced disease burden. Interestingly, as part of a larger effort to characterize cerebrospinal fluid from patients, researchers noted that this patient exhibited a significant enrichment of plasma cells and increased TCR clonality, suggesting that TCRs might be derived from such treated patients, potentially even T cell-excluded glioma patients. Results from a trial with an IDHR132H-mutant peptide vaccine similarly suggested the opportunity to identify therapy-driven functional TCRs. Bunse then described a phase 1 clinical trial in which 15 patients with newly diagnosed glioblastoma were treated with standard of care and a pre-manufactured tumor-associated antigen vaccine, and then later with a personalized neoantigen-specific vaccine. In this study, 4 out of the 15 patients were vaccinated with a distinct un-mutated epitope of the protein tyrosine phosphatase receptor type Z1 (PTPRZ1). PTPRZ1 is a glioma stem cell marker that is strongly overexpressed in various glioblastoma entities, and contributes to cancer cell proliferation, migration, and invasiveness. The immunogenicity of the PTPRZ11814-1822 antigen epitope was confirmed in all 4 patients, and 2 of these patients had partial responses. Bunse and his team were able to identify a PTPRZ1-reactive TCR from post-vaccination PBMCs obtained from one of the responding patients. A murinized version of this TCR was generated to avoid mispairing with endogenous human TCRs upon transfection of T cells. Co-culture assays confirmed the recognition of endogenously processed and presented PTPRZ1 and lysis of primary glioblastoma cell lines in an HLA-A*02:01-restricted manner by PTPRZ1 TCR T cells. Glioblastomas consist of cells that are present in 4 major distinct cell states. To understand whether treatment with PTPRZ1 TCR T cells would impact these cell states, individual patient tumor organoids were treated with PTPRZ1 TCR T cells. scRNAseq revealed that astrocyte (AC)-like and oligodendrocyte progenitor cell (OPC)-like glioblastoma cells, expressing higher levels of PTPRZ1, were eliminated first, followed by lysis of neural progenitor (NPC)-like and mesenchymal (MES)-like cells. In vivo, one dose of intravenous cells followed by two doses of intracerebroventricular PTPRZ1 TCR T cells improved radiographic responses and survival of mice with experimental intracerebral glioblastoma. In an effort to bring this novel off-the-shelf TCR into the clinic, Bunse and colleagues designed a clinical trial (INVENT4GB) in which up to 21 HLA-A*02:01-positive patients with glioblastoma will be treated with PTPRZ1 TCR T cells; the trial is planned to start in 2026. To modify the T cells, a non-viral, non-integrating, episomal scaffold matrix attachment region DNA vector encoding the TCR will be used. The T cell therapy will be first administered intravenously, before resection of the tumor and implantation of a catheter. The following three doses of PTPRZ1 TCR T cells will then be administered into the intraventricular space.
Novel tools to modify T cell recognition and tumor killing capacity - Sine Reker Hadrup, Technical University Denmark, Denmark
Sine Hadrup used Merkel cell carcinoma (MCC) to discuss novel methods to identify and boost antigen-specific T cells. MCC is a virally induced cancer driven by the polyoma large and small T-antigen oncogenes, and is sensitive to immunotherapy, despite a very low tumor mutation burden. Based on peptide binding predictions for Merkel cell polyoma virus peptides across 33 common HLAs, 1500 tetramers were prepared and used to screen T cells from checkpoint inhibitor-treated patients with MCC. Post-treatment T cells could be detected across multiple HLA alleles. Some patients had no recognition of MCC antigens, whereas others recognized up to 8 different T-antigen epitopes, with ~1% of the CD8+ T cells being MCC-specific in these patients. No T-antigen-specific T cell responses were detected in healthy donors, despite a high likelihood of having experienced viral exposure, and some detection of responses to the MCC capsid protein VP-1. Clinical outcomes were predicted by the levels of post-therapy responses to MCC T-antigens, but baseline responses were not predictive. An antigen tetramer-linked scaffold, also containing tethered cytokines IL-21 and IL-2, was used to expand antigen-specific T cells, leading to an increase in the number (~100x) and frequency of antigen-specific CD8+ T cells and a higher tumor killing capacity in vitro. The antigen scaffold-expanded T cell product may complement anti-PD-1 therapy to improve clinical outcomes. These scaffolds can also be used to expand and improve the fitness of TCR T cell or CAR T cell therapies. For example, following knock-in of CAR or TCR genes into the TRAC locus, the low frequency of successful knock-ins can be rapidly enriched by antigen/cytokine (IL-2, IL-7, and IL-15)-specific stimulation, and pooled scaffolds can be used to amplify multiple specificities simultaneously. The phenotype (CD27+CD28+), proliferative capacity, and in vivo killing capacity of the antigen scaffold-expanded TCR and CAR T cells could also be enhanced. Switching from a linear to a microparticle scaffold (T-Expand) further improved the ease of ex vivo cell expansion (as the biodegradable particles became internalized without toxicity, and will not have to be removed before transfer to the patient). When used instead of traditional CD3/CD28 bead-based stimulation, reduced inhibitory receptor expression, improved persistence, and better tumor control was observed. Delivery of T-expand in vivo was able to double the specific expansion of adoptively transferred OT-I cells. Finally, Hadrup described efforts to develop AI-enhanced, de novo-designed nanobinders to peptide:MHC, using NY-ESO-1 as an example. A set of 44 variants of designed nanobinders were tested, and only one bound tightly (6.8 nM), demonstrated acceptable specificity, and was cytotoxic in vitro in a CAR format. Initial efforts to target an A*01:01 neoepitope from a patient with melanoma are encouraging.
A pan-cancer atlas of T cell therapeutic targets - Guangyuan Li, New York University, USA
To expand the landscape of targets for T cell therapy, Guanyang Li presented ImmunoVerse, a tool to identify relevant intracellular targets presented by peptide:HLA (pHLA) to cognate TCR-engineered T cells. One strategy for identifying pHLAs utilizes conventional immunopeptidomics to isolate peptides bound to HLA, and determines the origin of such peptides by searching a database of the fragment masses derived from the native (or particular patient-specific mutated) proteome to identify matching patterns. However, such a database does not include many alternate neoantigen sources arising from the “dark matter” of the cancer genome, including alternative splicing, gene fusions, alternative small translation products, transposable element activation, and more. To address this limitation, Li developed ImmunoVerse, an expanded search database incorporating a broader array of such alternate peptide possibilities. Additionally, it has a stringent cut-off relative to normal tissue expression, and incorporates HLA binding prediction. Applied across 21 tumor samples, and incorporating many available omics datasets, these data revealed that in a canonical low-mutation-burden tumor type, such as AML, many potential neoantigens from dysregulated transposable elements and intron retention were observed. Overall, more than 27,000 tumor-specific peptides presented across multiple cancers were identified. One example of a novel epitope uncovered by this analysis was a peptide derived from HAVCR1, a gene important to and specifically expressed in renal cancer, and presented by the relatively common B*08:01 HLA allele. Generative AI was used to de novo design a binder to this pHLA complex, which, when engineered into a T cell, was shown to be specifically cytotoxic in vitro to two kidney cancer lines. Considering how ImmunoVerse might improve personal neoantigen cancer vaccines, overlapping of a large database (TSNAdb) of computationally predicted neoantigen peptides with the database of empirically identified peptides from ImmunoVerse revealed only a small overlap (0.001% of total epitopes in TSNAdb). However, Li argued that such epitopes should be prioritized based on empirical detection. One such (frame-shifted) peptide was identified in 16 different patients with MSI-driven colorectal cancer, and multiple other examples of recurrently detected peptides were observed. Li then described the interesting story of a transposable element, called long interspersed element-1 (LINE-1), which is epigenetically silenced in normal tissue, but is reactivated in some cancer cells, and has two open reading frames critical to the movement of this element in the human genome. The predicted protein from one of these two frames (ORF2p) could not be detected at the protein level by multiple assays. Hypothesizing that this protein was rapidly degraded by the proteasome, and therefore potentially efficiently presented by HLA, Li searched for peptides in the ImmunoVerse database, and found multiple ORF2p-derived presented peptides across different cancer types, with no detection in normal tissues. Finally, to round out the range of newly discovered targets, Li described detection of microprotein-derived tumor-specific antigens and a variety of viral and bacterial peptides derived from tumor-resident pathogens using the ImmunoVerse database as an enhanced search target. This database is available as an easily accessible web tool.
Cytokine therapies
Anti-cancer antibody-cytokine fusions: past, present and future - Dario Neri, Philogen/ETH Zurich, Switzerland
Conventional chemotherapy, and even monoclonal antibodies often exhibit limited tumor penetration and widespread distribution, leading to toxicity and reduced efficacy. Antibody fragments reach tumors faster (within 1 day) than full-sized antibodies (5 days), while small molecules demonstrate the most rapid (1 hour) and selective tumor accumulation in patients, achieving high tumor-to-background ratios within minutes. Dario Neri explored various strategies to improve the delivery of therapeutic payloads, such as radionuclides, cytokines, or cytotoxic drugs, in vivo. As a target, he picked the extra domain B (EDB) of fibronectin, which is largely absent in normal adult tissues, but highly and very frequently expressed in most tumors. Preclinical and clinical studies using nuclear medicine imaging proved preferential accumulation of the EDB-targeted antibody L19 in different tumor types, including primary and secondary brain tumors. Targeted delivery of cytokines to the tumor microenvironment could enhance local immune responses while minimizing systemic toxicity, and thereby could increase their therapeutic index more than 20-fold. Over the last 20 years, Neri and colleagues have extensively tested various disease-homing antibodies fused with a large number of different cytokine payloads. Some of these have been taken into the clinic, including L19–IL-2, L19–TNF, and L19–IL-12, among others. Preclinical studies revealed the distinct mode of action for each of these drugs. While systemically administered L19–TNF induced selective hemorrhagic necrosis of the tumor and acted as a “debulking” agent, L19–IL12 promoted an increase in intratumoral immune cells. Intralesional injection of L19–IL-2/L19–TNF (Daromun) in patients with melanoma showed high objective response rates in injected lesions, and even in distant, non-injected lesions, suggesting the induction of systemic antitumor immunity. This led to phase 3 clinical trials in the neoadjuvant setting for melanoma. Early results indicate a statistically significant prolongation of recurrence-free survival in patients treated with neoadjuvant L19–IL2/L19–TNF. Encouraging results have also been observed with intralesional administration of L19-IL-2/L19-TNF in basal cell and squamous cell carcinomas, avoiding the need for radical surgery. Further, Neri and his team are exploring systemic administration of L19-TNF (Fibromun) in heavily pre-treated patients with advanced soft tissue sarcomas and glioblastomas. Early phase 1 trial data suggest promising antitumor activity in patients when L19–TNF is combined with chemotherapy. As a next step, Neri and colleagues are focusing on developing strategies to further improve the therapeutic index of antibody-cytokine fusions. Shortly after infusion, the concentration of immunocytokines is high in the blood before the drug accumulates in the tumor and decreases in the blood, increasing the risk of adverse events in this early time frame. One approach to potentially alleviate this involves transiently inhibiting downstream signaling pathways of the delivered payload by co-administration of signaling inhibitors, such as TKIs, to mitigate early systemic side effects while maintaining long-term efficacy at the tumor site. Neri closed this talk by envisioning a future in which these smaller targeting agents may complement or even replace antibodies in certain therapeutic applications.
Lung-targeted cytokine-coding RNA-LPX for cancer immunotherapy - Ayline Kübler, TRON - Translational Oncology at the University Medical Center of the Johannes Gutenberg University, Germany
To improve lung cancer therapy for both primary and metastatic disease, Ayline Kübler described efforts to deliver cytokines directly to tumors. IL-2 and IFNα are both approved cytokine therapies, but are infrequently used due to systemic toxicity and short half-lives. By developing DOTMA/cholesterol-optimized lipoplexes (LPX) that directly target lung tissue and encapsulate cytokine-encoding mRNAs, Kübler hoped to overcome such deficiencies. To this end, they loaded LPXs with mRNAs encoding IFNα, a variant of IL-2 with low CD25-binding binding (to reduce toxicity) fused to albumin (IL-2alb; to enhance half-life), and IL-7 fused to albumin (IL-7alb). In a metastatic CT26 lung cancer model, i.v. administration of such RNA-LPX significantly extended survival, generated protective memory, increased proliferation and activation of CD8+ T cells and NK cells, and reduced Treg numbers and activation within the metastasis-bearing lungs. Single-cell RNAseq and differential gene expression analysis identified an upregulation of the type I IFN response signature, with enhanced NK and T cell effector functions (higher levels GrzA and GrzB), increased signs of activation, and a decrease in the phenotype of suppressive Tregs. Comparing various cytokine mixtures indicated the IL-2alb/IFNα/IL-7alb mix showed a strong synergistic effect, especially with regard to a higher CD8/Treg ratio. Depleting either CD8+ T cells or NK cells reduced the impact of the therapy, indicating that both cell types were required. In T cell escape models using a knockout of the dominant CT26 antigen (GP70) or of B2m, the cytokine-mix-encoding RNA-LPX treatment was still effective. However, additional knockout of NK cells or the activating NK cell receptor NKG2D eliminated the antitumoral effect of the RNA-LPX treatment in the CT26 B2m knockout model. These effects could be replicated in 3D-cultures of primary human non-small cell lung cancer treated with the supernatant of HEK293 cells that were lipofected with cytokine mix; this showed increases in activated, proliferating, granzyme B-expressing CD8+ T cells.
Cancer antigens
Revealing, Decoding and Targeting the endogenous cancer antigen repertoire - Nikolaos G. Sgourakis, University of Pennsylvania, USA
Most existing TCR-based therapies targeting HLA antigens are limited to a small subset of common HLA allotypes. In an effort to broaden the use of TCR-based therapies for more patients with different HLA allotypes, Nikolaos Sgourakis started out by investigating the crystal structure of a peptide derived from the public intracellular neuroblastoma oncofetal antigen PHOX2B bound to HLA-A24:02, and an scFv-based CAR (10LH) targeting PHOX2B/HLA-A24:02. While the CAR specifically recognizes arginine at position six of the peptide, its docking is also influenced by highly polymorphic framework residues on the HLA molecule, which are likely important clues to reactivity of other HLAs. By systematically perturbing these residues on the HLA-A24:04 backbone and monitoring the impact on binding, an empirical scoring system was developed to predict which of the 180 HLA allotypes could present the PHOX2B and be recognized by the 10LH CAR. This prediction was validated biochemically and through in vitro killing assays. Retrospective analysis of a pediatric cohort that was phenotyped at the Children’s Hospital in Philadelphia indicated that this therapy could potentially cover about 24% of patients, with the possibility of further expansion by targeting additional common HLA alleles like C*07:02, which cannot interact with the current CAR. This research highlights the concept of HLA cross-reactivity groups identified by the transplant community (e.g., the A9 serological group [CREG]). By analyzing shared patterns on the polymorphic surfaces of common HLA allotypes (A, B, and C), it is possible to classify HLA alleles into TCR cross-reactivity groups (T-CREGs). This classification, based on shared surface residues, can predict the restriction patterns of different TCRs and TCR-mimicking binders. An experimental assay using beads loaded with peptide:HLA complexes was developed to validate these predictions, showing that while individual TCRs often have narrow HLA restriction, the totality of TCRs can interact with all HLA allotypes. The inherent variability in the TCR's six CDR loops, and the ability to adopt diverse docking orientations on peptide:HLA complexes explain this broad potential for HLA interaction, complicating the development of tailored therapeutic TCR or antibody-based receptors. To overcome these limitations, a novel class of HLA-compatible binders called TRACeRs was engineered. The TRACeR platform features a rigid, computationally designed 25kd scaffold protein that docks on multiple of the highly oligomorphic HLA residues, with a minimalistic peptide recognition box comprising eight positions, which interact with the presented peptide across its length. Further, the TRACeR platform enables rapid generation of binders for different peptides presented by any specific HLA allotype (A, B, and C). This was demonstrated by the successful development of highly target-specific binders for multiple HLA-peptide combinations. TRACeR-based T cell engagers and CARs showed potent and specific in vitro killing of target cells, and minimal immunogenicity in mice. While covering all HLA allotypes for a specific antigen with traditional TCRs would require a vast number of unique receptors, TRACeRs offer a pathway to address broader patient populations. For instance, a TRACeR targeting PHOX2B in the context of the A9 serological group (similar to the existing 10LH CAR) showed on-target binding and killing. Remarkably, the same TRACeR scaffold could be readily reprogrammed to target PHOX2B presented by the HLA-C*07:02 allele, significantly expanding potential patient coverage.
Shared neoantigens’ atlas for off-the-shelf cancer vaccine development - Angela Mauriello, Istituto Nazionale Tumori - IRCCS Pascale, Italy
In a quest to create off-the-shelf neoantigen vaccines derived from common mutations and shared by different patients, Angela Mauriello looked at TCGA data to identify mutated proteins with single-nucleotide variants (SNVs) or short insertions or deletions (InDels) that were found in more than 5% of patients with a specific cancer type or shared by more than 5% of different cancer types. Subsequently, the NetMHCpan algorithm was used to study the putative neoantigenic epitopes stemming from the mutated proteins. To provide global coverage, binding to a total of 50 HLA alleles (10 HLA-A, 27 HLA-B, and 13 HLA-C) was examined. For HLA-A, HLA-B, and HLA-C, 11.7%, 9%, and 5.8% of the predicted neoantigens, respectively, were identified as strong binders, and the majority of these strong binders were predicted to bind to more than one haplotype. The ratio of HLA binding affinity (in nM) of the wild-type peptide divided by that of the mutated peptide (the differential aggretropicity index (DAI)) was used to define “true” neoantigens with a [DAI] of more than 10. For InDels, de novo peptide sequences without corresponding sequences in the wild-type protein were also considered as “true”, targetable neoantigens. According to this definition, 5 targetable SNV and 15 InDel neoantigens, 7 targetable SNV and 23 InDel neoantigens, and 0 targetable SNV and 10 InDel neoantigens were identified for HLA-A, HLA-B, and HLA-C, respectively. These neoantigens may have potential for clinical application. For example, the E545K mutation in PIK3CA was found in 7% of patients with prostate or bladder cancer, 19% of patients with breast cancer and 17% of patients with colorectal cancer. As some HLA haplotypes are more prevalent in certain global regions, particular mixtures of off-the-shelf neoantigen vaccines may be developed for a limited ethnic or regional group. In the next steps, Mauriello and her colleagues plan to experimentally validate the predicted shared neoantigens and prove their immunogenicity.
Bispecific antibodies
Precision targeting of intracellular proteins - Jan-Jaap Verhoef, Imuno Therapeutics, The Netherlands
Intracellular targets, which represent about 73% of the proteome, have so far mostly been out of reach for conventional immunotherapies, limiting their sphere of action to cell surface targets. In an effort to target intracellular disease antigens, Jan-Jaap Verhoef and his team set out to generate TCRmimic antibodies against peptide:HLA complexes. High-affinity binding often comes at the cost of specificity in engineered binders, like TCRs or antibodies, posing a bottleneck in therapy development. As specific drugs are crucial for patient safety, controlling for specificity should occur early in drug design. Interactions with the peptide mainly drive the specificity, while interactions with the HLA mainly determine the affinity. Screening tools, such as phage display libraries, are used to sift through billions of sequences, but often only focus on the affinity metric. Verhoef and team developed a pipeline combining different screening tools and structural mimicry to go through diverse antibody libraries, focusing on a balance of affinity and safety. Two programs initiated with this pipeline yielded high-affinity scFv binders (low nanomolar range) in single rounds. An identified TCRmimic antibody targeting a p53 neoantigen (R175H) was turned into a TCR-engager by combining it with a CD3-targeting domain; this was renamed IM810. The p53R175H driver mutation is carried by about 4-6% of patients with various cancer types, making it a relevant target. IM810 demonstrated high-affinity binding, with a Kd of 2.9 nM, and was highly potent against p53R175H-expressing HLA-A2+ cell lines, while no killing of p53WT HLA-A2+ cell lines was observed. In a search for potential cross-reactivity, Verhoef and his team applied the FDA-recommended assay X-scan, and identified 3 peptides that IM810 may bind to. In a highly sensitive recombinant antibody binding assay, IM810 demonstrated weak binding to these peptides complexed with HLA. However, testing with cell lines revealed that these peptides are not presented at the cell surface as pHLA complexes. The high-affinity and highly specific binders, identified with Verhoef’s pipeline, can be reformatted into potent T cell engagers, CARs, or ADCs.
Dual PD-L1 blockade and VEGF-A neutralization with the bispecific antibody BNT327/PM8002 shows potent antitumor activity in preclinical models - Maren Köhne, BioNTech SE, Germany
Maren Köhne described a series of pre-clinical experiments supporting the clinical translation of the PD-L1- and VEGF-A-targeting bispecific antibody BNT327. This single antibody has complementary modes of action, both preventing VEGF-A-induced abnormal vascularization and the subsequent formation of a suppressive tumor immune microenvironment, and blockade of the PD-1/PD-L1 axis to stimulate certain T cell populations to overcome exhaustion and enhance antitumor immunity. Both bivalent antibody domains bind their respective targets with sub-nanomolar affinity, and are active in relevant reporter assays with sub-nanomolar IC50's. Köhne and her colleagues co-cultured primary human CD8+ T cells engineered to express PD-1 and a CLDN6-targeting TCR with a human cancer cell line expressing CLDN6 and PD-L1. The addition of BNT327 to this cell mixture dose-dependently induced CD8+ T cells co-expressing CD107a and granzyme B, comparable to the anti-PD-L1 antibody atezolizumab, and caused tumor cell death, as measured over several days. Moreover, BNT327 dose-dependently increased IL-2 and IFNγ secretion in a mixed lymphocyte reaction assay. In a human PD-L1 knock-in Balb/c host inoculated with CT26 tumor cells engineered to express human PD-L1, mice with established tumors were B cell-depleted and treated with BNT327 or single agent-targeting antibodies. BNT327 outperformed the single-targeting antibodies in delaying tumor growth. In xenograft models (NOG mice supplemented with human PBMCs and implanted with various human tumor cell lines), BNT327 dose-dependently decreased tumor growth and outperformed single-agent, human PD-L1-, PD-1-, or VEGF-A-targeting antibodies. BNT327 has demonstrated safety and encouraging clinical results in combination with chemotherapy, and this combination is currently being evaluated in several phase II and III clinical trials. Furthermore, BNT327 is being tested in early-phase studies exploring its potential in combination with antibody–/drug conjugates.
By Ute Burkhardt and Ed Fritsch



