Aiming to improve clinical response to immunotherapy by better understanding the mechanisms behind the development of the heterogeneous population of exhausted CD8+ T cells (Tex), Beltra et al. extensively analyzed Tex cells in a chronic infection setting. The researchers identified four subsets with distinct transcriptional, epigenetic, and phenotypic features, as well as elucidated the key transcription factors that coordinated the development of and transitions between the subsets. The results were recently published in Immunity.
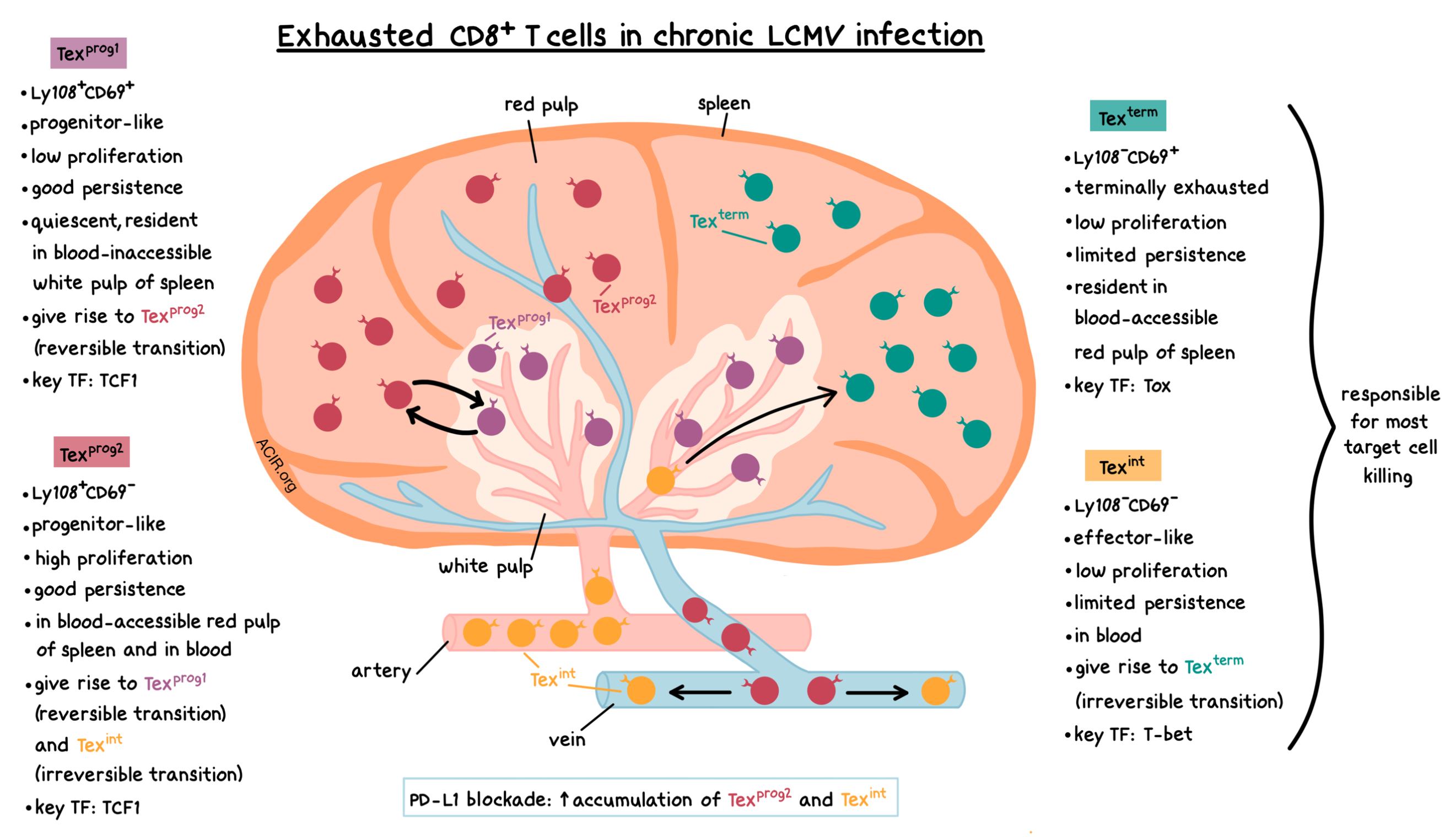
Beltra et al. began by adoptively transferring LCMV-specific CD8+ T cells into mice one day before infection with LCMV-clone-13 (chronic infection), and then analyzing the T cells 30 days post-infection. The researchers identified four subsets of exhausted (PD-1int or PD-1hi) CD8+ T cells (Tex) based on the expression of CD69 (an early activation marker that also plays a role in lymphocyte proliferation and egress) and Ly108 (encoded by Slamf6, and serving as a surrogate for the expression of TCF1 transcription factor). The identified four subsets were also sorted, separately adoptively transferred into infected mice, and analyzed after seven days. The observations from these two experiments showed the following characteristics of the four Tex subsets.
- Ly108+CD69+ (progenitor 1, Texprog1): This Tex subset had the highest TCF1 expression, good persistence, and low cell division, indicating its quiescent nature.
- Ly108+CD69- (progenitor 2, Texprog2): The cells in this Tex subset were TCF1+, highly proliferative, and had good persistence. Ly108+CD69- cells gave rise to Ly108+CD69+ cells and vice versa.
- Ly108-CD69- (intermediate, Texint): This Tex subset had limited persistence, but constituted a large fraction of Tex cells, even though these cells did not greatly proliferate themselves. This suggests that Ly108-CD69- cells arose from dividing Ly108+CD69- cells, and then lost further proliferative potential. Ly108-CD69- cells gave rise to Ly108-CD69+ cells.
- Ly108-CD69+ (terminal, Texterm): These cells exhibited limited persistence. They generally maintained their phenotype and did not proliferate. The phenotype of these cells was consistent with a PD-1hi terminally exhausted subset.
The four Tex subpopulations were present in chronic, but not acute, infection; were observed in multiple organs; and were also found in mouse B16 tumors and in TILs from human melanoma, indicating their conserved nature.
The researchers also noted several biomarker-related trends. Expression of CD69 was positively correlated with Tox and Eomes, and negatively correlated with proliferation and T-bet. Neither Ly108- subset gave rise to Ly108+ cells, which was consistent with the irreversible phenotype change upon the loss of TCF1. Thus, as the Tex cells progressed from Texprog1 to Texint, expression of Eomes, TCF1, and PD-1 decreased, while expression of T-bet increased. As the cells further transitioned to Texterm, the expression of T-bet decreased, while the expression of Eomes and PD-1 increased.
Next, Beltra et al. analyzed the location and function of the four Tex subsets. Texprog1 cells localized mostly in the blood-inaccessible white pulp of the spleen. Texprog2 cells were found in the blood-accessible red pulp of the spleen and in blood circulation. Texint cells were present in the blood as well. Texterm cells were not found in circulation, but were resident in the blood-accessible red pulp of the spleen and in other tissues. Texprog1 and Texprog2 cells showed few signs of cell death, while apoptosis was more frequently observed in the Texint and Texterm subsets, which was consistent with their limited persistence. Texprog1 cells produced the most cytokines (IFNγ, TNF), but Texterm cells showed the largest number of IFNγ/TNF co-producers. Texint and Texterm cells were responsible for the most target cell killing.
Exploring the transcriptional profiles of the four Tex subsets, RNAseq and gene ontology analyses revealed that Texprog1 and Texprog2 cells were similar to Tcf7+ progenitor-like cells, Texint cells exhibited an effector-like phenotype, and Texterm cells were identified as terminal Tex cells. Consistent with previously published data, Texterm cells had the highest expression of Tox. Thus, the transcriptional programs were consistent with the progressive developmental program of Tex cells and the acquisition of Tex subset-specific biological functions.
Using ATACseq, the researchers then explored the epigenetic relationships between the four Tex subsets. They observed epigenetic similarities between Texprog1 and Texprog2 subsets, but significant epigenetic remodeling during Texprog2 to Texint and Texint to Texterm transitions. The remodeling tracked with developmental observations: genes related to progenitor biology (e.g., Tcf7, Il7r) were epigenetically silenced during the Texprog2 to Texint transition and remained inaccessible in future transitions. During Texint to Texterm transition, effector genes (e.g. Cx3cr1) became less accessible and exhaustion-related genes (e.g. Cd160, Nr4a family members) became more accessible.
Combining ATACseq and RNAseq data together with knockout experiments revealed key transcription factors for different Tex subsets. TCF1 played a key role in Texprog1 and Texprog2 cell development. T-bet played an essential role in the development of the Texint population. Tox expression decreased as the Tex cells progressed from progenitor cells to Texint cells, but then increased again in Texterm cells. Tox expression negatively correlated with T-bet expression in Tex cells in both chronic infection and cancer, and experiments using adoptive co-transfer of WT and Tox+/- T cells demonstrated that Tox negatively regulated T-bet. Thus, Tox was a key regulator of the progression of Texint cells to Texterm cells due to its ability to control T-bet expression in Tex cells.
Finally, tying this data to clinical relevance, the researchers adoptively transferred LCMV-specific CD8+ T cells into mice, infected them with LCMV, and treated them with anti-PD-L1. PD-L1 blockade led to the preferential accumulation of Texprog2 and Texint cells one day post-treatment, and this was at least partially dependent on CD4+ T cell help. Thus, PD-1 blockade may preferentially act on these two Tex subsets, and could lead to the conversion of Texprog2 cells into Texint cells.
In this detailed study, Beltra et al. characterized four subsets of exhausted CD8+ T cells present within a chronic infection setting, with distinct transcriptional, epigenetic, and phenotypic features. The authors also identified TCF1, T-bet, and Tox as the key transcription factors that coordinated the development of and transitions between the Tex subsets, and demonstrated that PD-L1 blockade preferentially acts on two of the subsets. Given that the Tex subsets were also found in mouse and human tumors, elucidating these details may prove relevant for improving the design and clinical outcomes of immunotherapy.
by Anna Scherer
Meet the Researcher
This week, first author Jean-Christophe Beltra answered our questions.

What prompted you to tackle this research question?
CD8+ T cell exhaustion is really a central process to critical diseases such as chronic viral infections, cancers, and autoimmune disorders, and trying to prevent and/or reverse this process is a key feature of the most promising anti-cancer immunotherapies such as CAR T cell infusion or immune checkpoint blockade. In regard to that, it really appeared paradoxical to me that we still ignore a lot of the developmental biology of these cells. It is particularly relevant currently as combinatorial approaches, such as combining immune checkpoint inhibitors such as anti-PD-1 or anti-CTLA-4 with other immuno-regulatory agents, provide significantly better outcomes in preclinical models and patients. Yet, how to efficiently manipulate exhausted CD8+ T cells with multiple agents, how to pick the magic combination that will achieve the best possible response in patients, starts with a better comprehension of the intimate biology of these cells.
What was the most surprising finding of this study for you?
One interesting perspective I like about this study is the observation that blocking the PD-1/PD-L1 pathways may provide its known clinical benefits at least in part by accelerating and intensifying a naturally existing process that is a full part of the developmental biology of exhausted CD8+ T cells. Indeed, we observed that at some point in their development, exhausted CD8+ T cells downregulate PD-1 expression, and this biological event coincides with a regain of proliferation and effector biology, which are known outcomes of PD-1/PD-L1 blockade therapy. The initial reasoning behind targeting the PD-1 receptor was its intense and selective expression by exhausted CD8+ T cells. It is pretty clear from several past studies that PD-1 acts as a biological brake for CD8+ T cell responses to chronic antigen stimulation, but what our study suggests is that this brake is temporary and partially released during the natural developmental process of exhaustion, allowing these cells to amplify and fight the infection. Now that we know about this process, we might be able to define other pathways involved that also support this regain of proliferation and effector biology in exhausted CD8+ T cells, and use this knowledge to provide these cells with the boost they need to fight cancer, particularly in the context of PD-1/PD-L1 blockade.
What was the coolest thing you’ve learned (about) recently outside of work?
As a young father, I just can’t stop talking about my little guy. It’s really fascinating for me to see how much he understands day after day about his surroundings and how he is putting information together to make a new big step. Learning the sound of vowels helped him to pronounce new words, and learning to balance his body helped him stand and make his first steps. I found it extremely funny to think that as a grown-up adult, I am going through the exact same process every day at work, putting pieces of information together to help solve the gigantic puzzle of immunological processes.



