Given that only a subset of patients responds to current checkpoint inhibition therapies, the search for new immune checkpoint targets continues. Sharma et al. identified and investigated one such new target, LILRB4, in mouse and human tumors, and assessed the effects of blockade in a variety of mouse models. Their results were recently published in the Journal of Experimental Medicine.
The researchers started by investigating tumors from the B16F10 mouse melanoma model using Nanostring RNA analysis. They found high expression of Lilrb4 and Cd276 (PD-L1) as the two most highly expressed genes. LILRB4 was found by flow cytometry on CD45+ TILs from the B16F10 tumor, as well as in TILs in other mouse models, while it was absent from the spleen. LILRB4 expression was also high in tumor tissue from melanoma patients, and TCGA data showed that tumors had higher ratios of LILRB4/CD3ε mRNA than healthy tissue in breast cancer, lung squamous cell carcinoma, and lung adenocarcinoma.
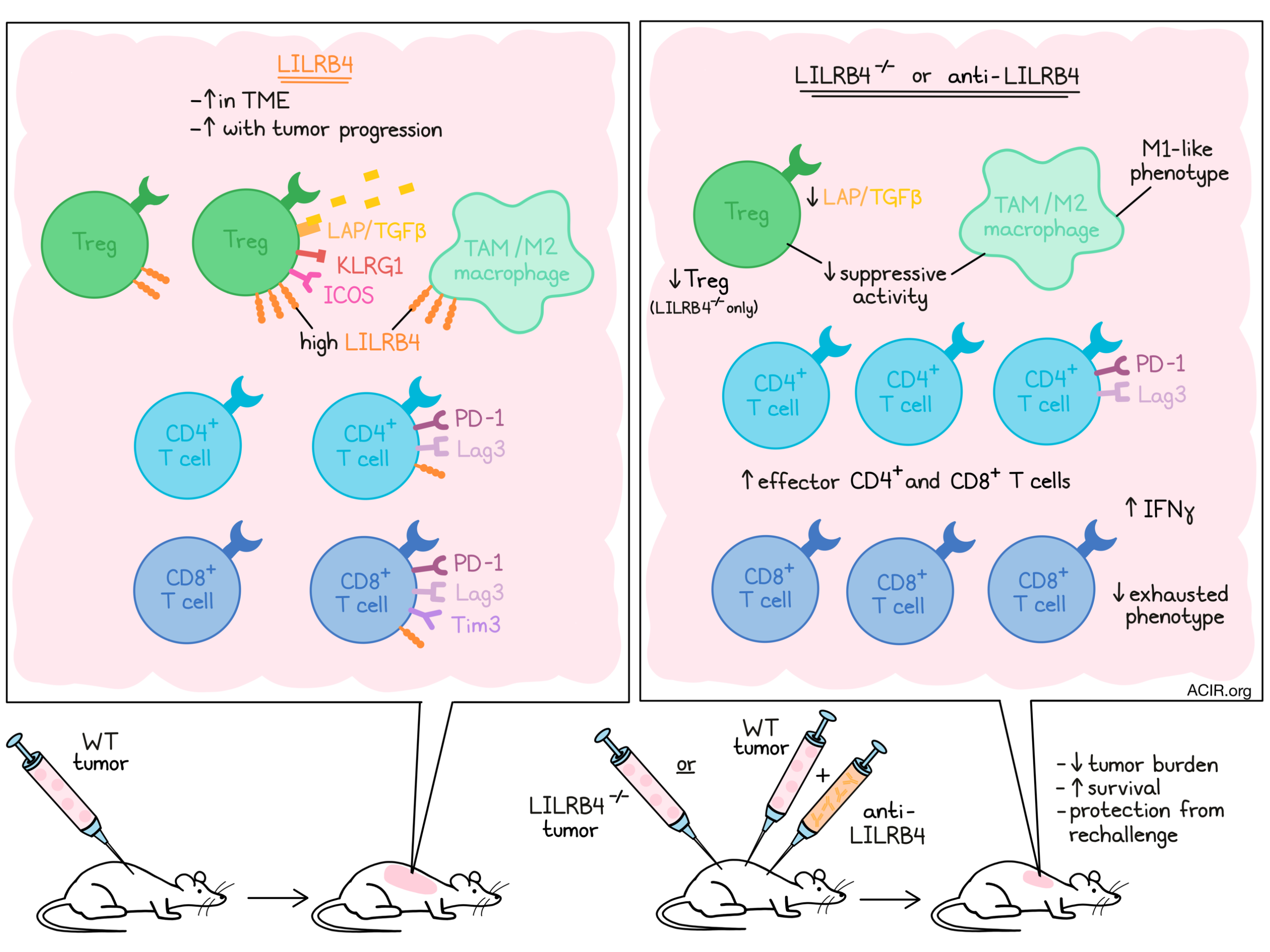
Evaluating which immune populations expressed LILRB4 in mouse tumors, the researchers found that CD3+ T cells, CD11b+ F4/80+ macrophages, and CD11b+GR-1+ neutrophils expressed the highest levels, while DCs, NK cells, and NKT cells expressed low levels, and B cells were negative. Within the T cells, LILRB4 expression was highest on CD4+ T cells, in particular on Tregs, and increased upon tumor progression.
To further establish the immune populations expressing LILRB4, the researchers performed mass cytometry with more than 25 markers on cells obtained from MC38 and B16F10 tumors, followed by unsupervised clustering and assignment of cell type to each cluster. LILRB4 was expressed highest on Tregs, with the highest expression in the KLRG1highICOShighTGFβhigh cluster. Among CD4+ T effector cells, expression was higher on the PD-1highLag3high cluster, and on CD8+ T cells it was only expressed on the PD-1highLag3highTim3high cluster. These data suggest LILRB4 is expressed mainly on Tregs and on “exhausted” T cells that express other inhibitory receptors. Confirming these findings in TCGA data, LILRB4 gene expression was correlated with various functional molecules, such as granzymes, perforin, and IFNG, and with inhibitory molecules, such as PD-1 and Tim3, in multiple tumor types.
To assess LILRB4 on myeloid cells, Sharma et al. deployed a myeloid CyTOF panel on MC38 tumors. CyTOF analysis revealed LILRB4 expression on all monocyte/macrophage clusters, but to a variable extent. The highest expression was in clusters expressing the immunosuppressive tumor-associated macrophage (TAM) markers CD206, Arg-1, CX3CR1, CD163, and IDO. Analysis of in vitro-skewed M1 and M2 bone marrow-derived macrophages (BMDM) confirmed the higher expression of LILRB4 on M2-skewed macrophages.
Next, TIL from MC38 tumors were subjected to scRNAseq and clustered. Again, Lilrb4 was mainly found on CD3+ T, CD11b+, and CD11c+ cell clusters. Expression on monocytes/macrophages and neutrophil clusters was highest, and macrophage clusters expressing markers associated with M2 macrophages had the highest expression of Lilrb4.
Sharma et al. then moved to test the effects of blockade of LILRB4 using a polyclonal antibody in the B16F10 model. The antibody was injected intratumorally after tumor inoculation, and mice receiving this antibody had decreased tumor burden and increased survival as compared to control mice. Similarly, LILRB4-/- mice had lower tumor burden and longer survival after B16F10 or mT5 (pancreatic tumor model) tumor challenge than WT mice.
Based on the findings, the researchers developed a monoclonal hamster anti-LILRB4 antibody and treated mice with MC38 tumors by intraperitoneal injection, which improved their survival. Assessing whether the response induced memory, surviving mice were rechallenged with tumor cells without additional treatment. All mice cleared the tumors and survived, suggesting the antibody induced tumor-specific immunity.
Looking into the tumor microenvironment of B16F10 tumors grown in LILRB4-/- and WT mice, Sharma et al. found that genes such as Cd3e, Cd8a, and GZMB were upregulated in the tumors from LILRB4-/- mice. These findings were confirmed by assessing TILs in MC38 tumors, where the mutant mice had increases in CD3+, CD8+, and CD4+ effector T cells, and a decrease in Tregs. In WT mice treated with the LILRB4 antibody, there was also a significant increase in CD8+ and CD4+ effector T cells, and a decrease in the frequency of Tregs.
To determine the suppressive effects of Tregs, T cells were isolated from the spleens of WT and LILRB4-/- mice, and incubated with naive T cells, which were activated using CD3 and CD28 antibodies. In the presence of the WT Tregs, the CD8+ and CD4+ T cells produced less IFNγ than those in the presence of LILRB4-/- Tregs. Given that TGFβ is one mechanism by which Tregs can suppress effector cell function, the level of the latent LAP/TGFβ complex (precursor to active TGFβ) was assessed on both Treg subsets. The LILRB4-/- Tregs showed lower surface expression of LAP/TGFβ than the WT Tregs, suggesting this is a potential mechanism of suppression.
The suppressive effects of macrophages under the influence of LILRB4 were analyzed in a similar way. Bone marrow-derived macrophages from WT and LILRB4-/- mice were skewed in vitro to the M2-phenotype using IL-4 and co-cultured with activated T cells. In the presence of the LILRB4-/- macrophages, the T cells secreted more IFNγ. Comparing the expression of cytokine genes suggested that the LILRB4-/- M2-skewed macrophages had an increased expression of IL1b and Nos2, suggesting these cells had a more inflammatory M1-like phenotype.
Finally, the infiltration of myeloid and lymphoid cell subsets in tumors treated with the anti-LILRB4 antibody was assessed using CyTOF. Macrophage clusters expressing Arg-1, CV3CR1, and IDO decreased, suggesting less suppressive activity, while monocyte/macrophage clusters without suppressive phenotypes increased after treatment. No significant changes in Treg clusters were detected, but the CD4+ Th1 effector cell cluster, which did not express inhibitory receptors such as PD-1 and Tim3, was increased in frequency after antibody treatment. Among CD8+ T cells, the cluster expressing PD-1, Lag3, and Tim3 showed a decrease after treatment.
The data presented in this study suggest that LILRB4 is an immunosuppressive target on tumor-associated Tregs and macrophages that can be modulated to decrease immune suppression in the tumor microenvironment. If these results can be confirmed in patients, the next question of interest is to assess whether anti-LILRB4 treatment works synergistically with checkpoint blockade to overcome some of the immune barriers in non-responders.
Write-up by Maartje Wouters, image by Lauren Hitchings
MEET THE RESEARCHER
This week, first author Naveen Sharma answered our questions.
What prompted you to tackle this research question?
Many cancers are refractory to current immunotherapy drugs. Therefore, there is an ongoing effort to find new immunotherapy targets and implement new combination therapies. Immunosuppressive tumor-associated macrophages (TAMs) constitute a large fraction of the tumor microenvironment (TME) and express several immune inhibitory receptors, which can be targeted to modulate their function from pro-tumor to anti-tumor. We were interested in identifying inhibitory receptors that modulate the intratumoral macrophage–T cell relationship and could be targeted for tumor immunotherapy.
What was the most surprising finding of this study for you?
In our study, we found that LILRB4 was expressed at relatively higher levels on pro-tumor M2-type macrophages and Tregs. Interestingly, bone marrow-derived M2-skewed macrophages and Tregs that lack LILRB4 were less suppressive compared to the wild-type (WT) counterparts in in vitro T cell functional assays. This was a surprising finding that the lack of LILRB4 inhibitory receptors on these cell types inhibited their suppressive function. These results suggest that the LILRB4 receptor on these cell types could engage some unknown LILRB4 ligand on T cells to reduce their function.
What was the coolest thing you’ve learned (about) recently outside of work?
One of the coolest things I learned recently is fossil hunting as a hobby. I recently went to a fossil digging site with my six-year-old son, who is very interested in dinosaurs and fossils. This site allows amateurs to see and dig fossils, and one can also keep them for personal use. These fossils were from the ”Pennsylvanian Period” and were approximately 300 million years old. It was a fun learning experience and was also a reminder of the recent evolution of humans compared to the evolutionary timescale of life on earth and how humans have such a significant impact on the environment in such a short duration of existence.



