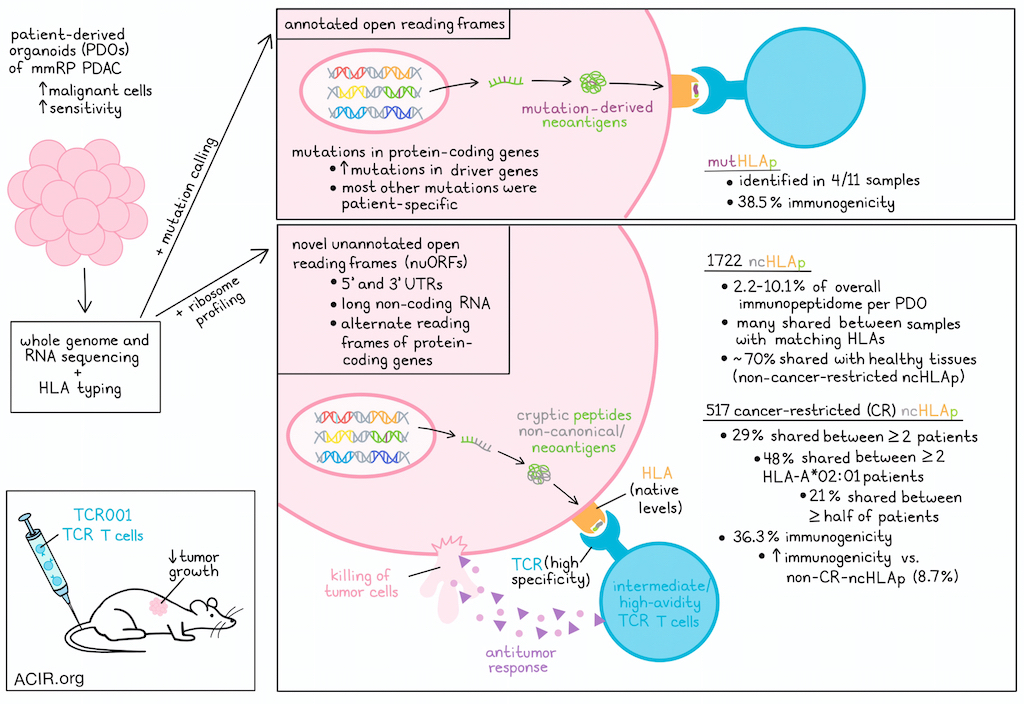
Pancreatic ductal adenocarcinoma (PDAC) has proven resistant to immunotherapy strategies, in part due to limited tumor mutational burden and T cell infiltration. Besides tumor-specific somatic mutations resulting in neoantigens, cancer cells can also translate genome regions outside of annotated open reading frames (ORFs), resulting in HLA-I presentation of cryptic antigens derived from regions such as 5’ and 3’ untranslated regions (UTRs), long noncoding RNAs (lncRNAs), or alternative reading frames (intORFs) of protein-coding genes. Searching for targetable antigens, Ely, Kulstad, et al. used immunopeptidomics to investigate these non-canonical HLA-I-bound peptides (ncHLAp) in PDAC, and assessed their immunogenicity and cancer specificity in work recently published in Science.
Since PDAC generally has low malignant cellularity (20-30%), the researchers established patient-derived organoids (PDOs) from mismatch repair-proficient PDAC, hypothesizing that this would enrich malignant cells and improve sensitivity. The PDOs and paired normal tissue were subjected to whole-genome and RNA sequencing for mutation calling and HLA typing. For some patients, autologous bulk tumors were subjected to whole-exome and RNAseq. Comparing these bulk samples with the PDOs showed lower expression of stromal and immune gene signatures and improved sensitivity for mutation detection of the PDOs.
Twelve PDOs were selected for further characterization. The most frequent mutations were detected in PDAC driver genes, but most other somatic mutations were patient-specific. Eleven of the PDOs had intact HLA-I expression and were responsive to IFNγ. The immunopeptidomes of these 11 PDOs were profiled using liquid chromatography-tandem mass spectrometry. This approach identified >91,000 distinct HLAp, with 8,000-18,500 per patient sample, and most were 8-11 amino acids in length, as expected. To assess whether use of PDOs indeed selected for peptides more specific to cancer cells, HLA-I immunopeptidomics was performed on three unmatched primary PDAC bulk tumor samples, and empirically identified HLAp were mapped to the canonical source genes to define a peptide source gene module (PSGM) for each bulk tumor or PDO sample. Comparing scRNAseq of bulk tumor samples and previously published scRNAseq data with these data revealed that the PDO-derived PSGMs were enriched in malignant cells of the bulk tumor samples, while the bulk sample-derived PSGMs were enriched in non-malignant cell populations.
For comparison, Ely, Kulstad, et al. searched for peptides derived from patient-specific mutations, including germline variants, in each PDO sample, and found 5 endogenously expressed mutation-derived neoepitopes (mutHLAp) across 4 specimens (37%). In contrast, the remaining 7 specimens (63%) had none, suggesting only a small percentage of non-synonymous exonic mutations produced mutHLAp.
To identify ncHLAp in PDAC, the immunopeptidomic search was focused on novel unannotated ORFs (nuORFs) identified through ribosome profiling. This detected 1,722 ncHLAp across 11 PDOs, which accounted for 2.2-10.1% of the overall immunopeptidome per PDO. Many of these ncHLAp were shared among PDOs with matching HLA, and >50 were detected in ≥2 HLA-A*02:01, HLA-B*44:02, or HLA-C*05:01 samples.
To determine which of the detected ncHLAp were cancer-specific, the researchers obtained ncHLAp from immunopeptidomic data from 28 healthy tissues, ribosome profiling of 8 healthy tissues, and immunopeptidomics of healthy thymus. Healthy tissues had large numbers of ncHLAp, with almost 70% of the identified ncHLAp in PDOs also detected in some healthy tissues. The healthy tissue ncHLAp were filtered from the PDO-derived ncHLAp to identify cancer-restricted (CR) ncHLAp, which identified 517 (30.2%) CR ncHLAp. Of these, 29% were shared between two or more patients, and 48% of the HLA-A*02:01 CR ncHLAp were shared, with 21% shared between over half of HLA-A*02:01+ patients.
To determine whether ncHLAp can be recognized by T cells, an ex vivo platform was used with HLA-matched healthy donor PBMC-derived DCs to prime and expand autologous CD8+ T cells. Of the CR ncHLAp tested in this system, 36.3% generated antigen-specific T cells that were undetectable in precursor populations. The CR ncHLAp had a higher immunogenicity rate (36.3%) than non-CR ncHLAp (8.7%). When mutation-derived neoepitopes were assessed using the same platform, a similar proportion demonstrated immunogenicity (38.5%) as the CR ncHLAp.
Next, the researchers determined whether ncHLAp-reactive TCRs could recognize endogenous levels of CR ncHLAp on cancer cells. The 10X genomics barcode-enabled antigen mapping workflow was combined with the ex vivo T cell priming and expansion platform to identify candidate TCRs. This identified 17 candidate CR ncHLAp-reactive TCRαβ clonotypes. TCR-T cells were engineered with the detected TCRs using CRISPR-Cas9 to knockout the endogenous TCRαβ, and lentivirus to introduce the identified TCRs. The functional avidity of the TCR-T cells was tested through co-culture with TAP-deficient T2 cells loaded with the peptides. Three candidates exhibited very low, two high, and one intermediate functional avidity. The TCR-T cells were then co-cultured with the PDOs to assess whether they could detect endogenous levels of presented peptides. While the three low-avidity TCR-T cells did not recognize cognate antigens, the other three TCR-T cells demonstrated antigen recognition, produced effector cytokines, and upregulated 4-1BB (CD137). Further, each TCR-T induced killing of PDO cells, even at low effector:target ratios.
The peptide (NU11) recognized by the most potent ncHLAp-directed TCR (TCR001) was further characterized by alanine scanning to dissect the residues of NU11 critical for recognition by the TCR. This revealed that any substitutions within NU11 removed TCR001 recognition. ScanProSite was then used to search for other human peptides with possible cross-reactivity, but none were detected, further confirming its high specificity.
Finally, the researchers assessed the efficacy of the ncHLAp-directed TCR-T cells in inhibiting tumor growth in vivo. PDAC PDOs were subcutaneously transplanted into athymic mice, and even a single administration of 1x107 TCR001 TCR-T cells resulted in tumor progression delay, while two other high-avidity TCRs did not inhibit growth. However, all tumors eventually progressed. In NSG mice, two administrations of the TCR001 TCR-T cells significantly delayed tumor progression. Improvements in production, function, and persistence of the TCR-T cells would be expected to further enhance antitumor effects.
These data present a novel platform to uncover a new source of neoantigens that are cancer-specific and immunogenic, even in a cancer type with a low mutational burden. Identification of this antigen class and the finding that these antigens can be targeted using TCR-T cells provides additional avenues for the development of vaccine and TCR-T cell therapies.
Write-up by Maartje Wouters, image by Lauren Hitchings
Meet the researcher
This week, lead author William Freed-Pastor answered our questions.
What was the most surprising finding of this study for you?
We set out to empirically profile the antigen landscape in pancreatic cancer using mass spectrometry-based immunopeptidomics. While we were able to identify some endogenously expressed mutation-derived neoepitopes, what was quite surprising to us was the sheer abundance of non-canonical (or cryptic) HLA-I bound peptides in the immunopeptidome. These had been described in multiple other tumor types, but never before in pancreatic cancer. We were also surprised by the fact that these cryptic antigens were so robustly immunogenic and could directly mediate tumor cell killing when using T cell receptor (TCR)-engineered T cells.
What is the outlook?
We now have multiple ongoing projects in the laboratory investigating the mechanistic basis for aberrant translation in pancreatic cancer, expanding our antigen discovery work into other classes of potential antigens, and leveraging our TCR discovery pipeline to nominate the most effective TCRs to target this class of antigens. We are actively working with colleagues in the Hale Family Center for Pancreatic Cancer Research at Dana-Farber Cancer Institute to translate these basic science discoveries into a clinical trial, which we hope will launch in the next two to three years.
If you could go back in time and give your early-career self one piece of advice for navigating a scientific career, what would it be?
One piece of advice that I received early on in my career was to follow the science wherever it leads (perhaps especially when it takes you in unexpected directions). I think that this has been an excellent piece of advice and has proven to be a great Northern Star. Another piece of advice is don't be afraid to tackle challenging questions. If we really want to see transformational change for cancer patients, we need to be willing to invest the time and energy (both individually and collectively) to tackle the most difficult and pressing questions.



