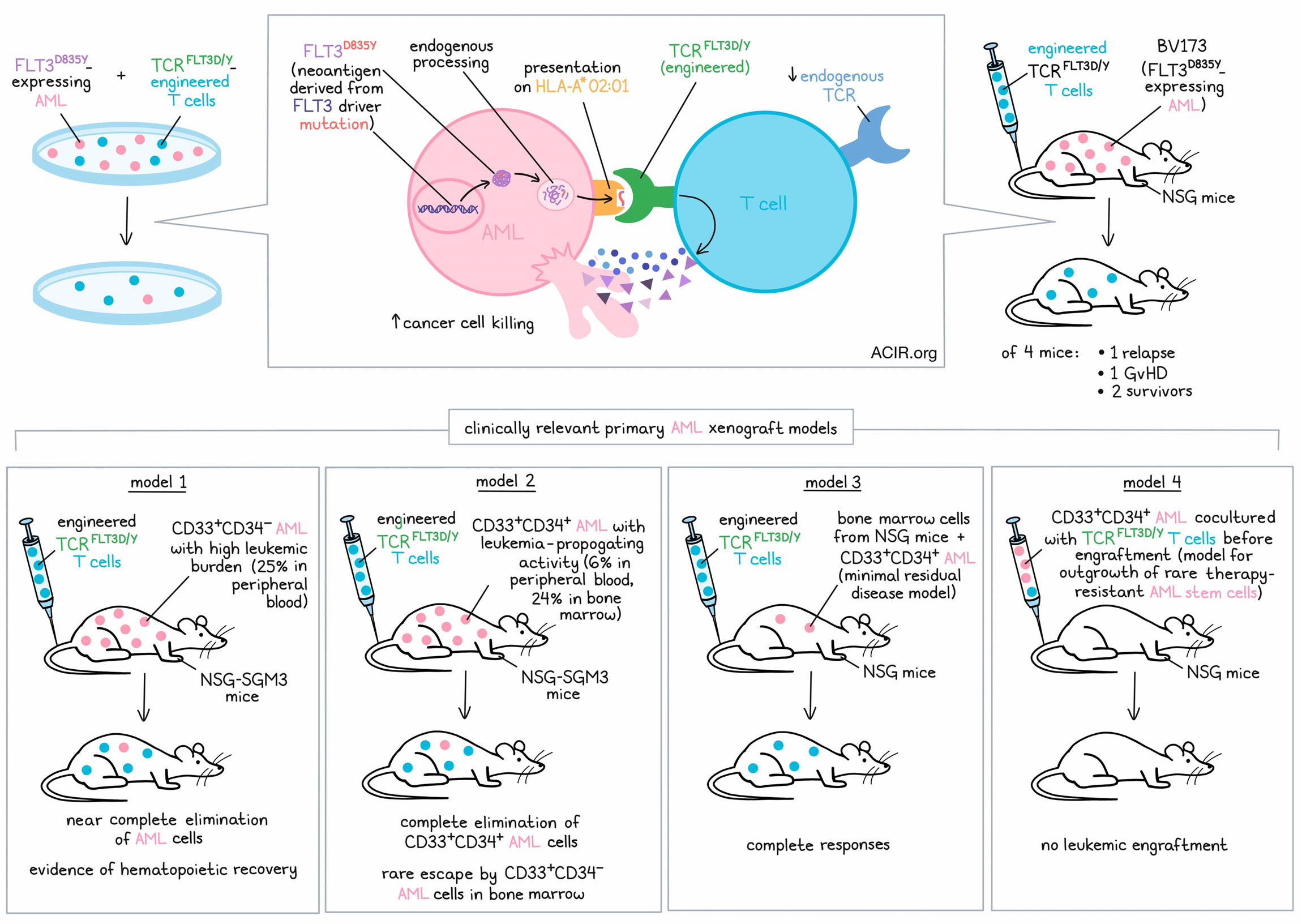
Acute myeloid leukemia (AML) is the most frequent leukemia in adults. Currently, allogeneic hematopoietic stem cell transplantation, although associated with high risks for relapse, transplant-related morbidity, and graft-versus-host disease, is the only curative treatment option. At present, CAR T cell therapies targeting myeloid cell surface antigens that are overexpressed in AML, but are also expressed on normal myeloid progenitor cells and/or hematopoietic stem cells, are being cautiously studied. AML is driven by recurrent mutations in a restricted number of genes. However, the resulting shared neoantigens are inaccessible to CARs. In a recent study published in Nature Cancer, Giannakopoulou et al. demonstrated the therapeutic potential of T cells engineered to carry a TCR with specificity for a mutation in FTL3, a recurrent driver in AML.
Mutations, particularly D to Y, at position D835 in FLT3 are found in 7-10% of patients with AML and represent the most frequent mutations in FLT3. Reanalysis of past DNA sequencing data of AML patients carrying the FLT3D835Y mutation revealed that FLT3D835Y also had the highest variant allele frequency (VAF) among identified recurrent driver mutations, or was secondary only to clonal hematopoiesis mutations, indicating that FLT3D835Y might be an AML-initiating or transforming mutation.
To identify TCRs reactive to HLA-A*02:01-restricted FLT3D835Y, Giannakopoulou and colleagues screened naive T cells from 16 healthy blood donors upon co-culture with autologous monocyte-derived DCs electroporated with mRNA encoding a FLT3D835Y minigene encompassing two 9- and 10-mer predicted mutant binding peptides. CD8+ T cells with reactivity to a FLT3D835Y 9-mer were detected in only one donor (no reactivity to the 10-mer peptide was detected), and analysis of 55 single cells resulted in the identification of only one functional TCR sequence (TCRFLT3D/Y). Immunopeptidomics and targeted mass spectrometry of primary AML cells obtained from two patients confirmed endogenous processing and presentation of the FLT3D835Y 9-mer in AML. The FLT3D835Y 9-mer complexed with HLA-A*02:01 demonstrated an almost 10-fold higher half-life than the wild-type peptide, suggesting strong immunogenicity of the neoantigen.
After introduction of murine constant regions into the TCR and retroviral transduction, TCRFLT3D/Y was efficiently expressed in healthy donor peripheral blood T cells. Analyses revealed preferential pairing of the introduced TCR-α and -β chains, suppression of the endogenous TCR, and a mainly naive profile of transduced CD4+ and CD8+ T cells. TCRFLT3D/Y cells recognized picomolar concentrations of FLT3D835Y 9-mer on K562 target cells with a half-maximal effective concentration (EC50) of 81 pM, but did not recognize the corresponding wild-type peptide. Mapping of the fine specificity of TCRFLT3D/Y by saturation mutagenesis of the 9-mer peptide identified 28 additional 9-mer peptides in the human proteome that TCRFLT3D/Y could potentially recognize. Giannakopoulou and colleagues, however, found that TCRFLT3D/Y reacted to only three of these peptides. All three peptides were derived from proteins with ubiquitous expression (LZTR1, MED1, and PRADC1). Since TCRFLT3D/Y cells showed no or negligible reactivity to a panel of 26 HLA-A*02:01+ cell lines of different tissue origins and HLA-A*02:01+ K562 cells electroporated with mRNA encoding peptides derived from LZTR1, MED1, and PRADC1, the authors concluded that these cross-reactive peptides were not naturally processed and presented.
TCRFLT3D/Y cells efficiently killed a mean of 87% of all myeloid cells from seven patients with HLA-A*02:01+ FLT3D835Y AML at a low effector:target ratio of 1:2, while CD3+ T cells and CD19+CD20+ B cells were not affected. At the same time, TCRFLT3D/Y cells did not kill FLT3D835E, FLT3D835H, or FLT3wild-type HLA-A*02:01+ cells, nor did they kill FLT3D835Y HLA-A*02:01- patient cells, confirming specificity and HLA-A2 restriction. When leukemia cell lines were transduced to express FLT3D835Y, TCRFLT3D/Y cells were able to kill more than 95% of these cells within 24 hours at an effector:target ratio of 1:2. Moving closer to the clinical setting, Giannakopoulou and colleagues transduced T cells obtained from patients with AML and demonstrated specific and efficient killing of autologous AML cells by the patient-derived TCRFLT3D/Y cells.
When NSG mice engrafted with the B cell precursor leukemia cell line BV173, transduced to express FLT3D835Y, were treated with TCRFLT3D/Y cells, leukemic cells were undetectable by bioluminescence imaging at day 21, while control mice had to be sacrificed due to high leukemic burden at the same time point. One of the four treated mice developed graft-versus-host disease, another mouse had a relapse on day 28, and the two others survived. Analysis of the bone marrow of the surviving mice revealed no leukemia cells but persistent TCRFLT3D/Y cells at the end of the experiment on day 53.
Finally, Giannakopoulou and colleagues analyzed the in vivo efficacy of TCRFLT3D/Y cells in four different clinically relevant primary AML xenograft models:
Model 1: CD33+CD34- AML with high leukemic burden
NSG-SGM3 mice (known to specifically enhance human myeloid lineages) were highly engrafted with primary FLT3D835Y CD33+CD34- AML just prior to TCRFLT3D/Y cell infusion, with human CD33+ cells making up about 25% of the peripheral blood cells. Following infusion with TCRFLT3D/Y, virtually all leukemic cells were eliminated by day 14. Droplet digital PCR confirmed an almost complete elimination of the human CD33+ cells in the BM, while the number of murine CD45+ cells in bone marrow increased, indicating recovery of mouse hematopoiesis.
Model 2: CD33+CD34+ AML with leukemia-propagating (“AML stem cell”) activity
NSG-SGM3 mice were engrafted with primary FLT3D835Y CD33+CD34+ AML, with human CD33+ cells occupying about 6% of the peripheral blood cells and about 24% of the bone marrow cells. At the end of the experiment on day 34, CD33+CD34+ cells were completely eliminated in the bone marrow of mice treated with TCRFLT3D/Y cells, but some CD33+CD34- cells (mostly without the FLT3D835Y-mutation) persisted in the bone marrow, although at greatly reduced levels compared to control mice.
Model 3: Minimal residual disease
Bone marrow cells from NSG mice engrafted with primary CD33+CD34+ AML were used to engraft another set of NSG mice (secondary transplantation), resulting in a low, but stable leukemia burden. Treatment with TCRFLT3D/Y cells resulted in a complete elimination of the primary AML cells.
Model 4: Outgrowth of rare, therapy-resistant AML stem cells
To model a scenario in which rare, therapy-resistant AML stem cells outgrow in vivo, primary CD33+CD34+ AML cells were cultured with TCRFLT3D/Y cells in vitro for 48 hours before transplantation into NSG mice. Whereas mice treated with control AML cells showed progressive and high leukemic engraftment, no leukemic engraftment was detected in mice transplanted with AML cells co-cultured with TCRFLT3D/Y cells. Of note, the few TCRFLT3D/Y cells transplanted with the AML cells did not persist in the mice either.
Here, Giannakopoulou and colleagues describe a new potential treatment option for AML with TCR-engineered T cells targeting a recurrent driver mutation in FLT3. In primary AML xenograft models of different clinical scenarios, the researchers provide proof that their TCRFLT3D/Y cells can eliminate the reservoir of AML stem cells by targeting a single shared neoantigen. 3-4% of patients with AML express the mutation and HLA-A*02:01, and could potentially benefit from the treatment.
Write-up by Ute Burkhardt, image by Lauren Hitchings
Meet the researcher
This week, lead author Johanna Olweus answered our questions.

What was the most surprising finding of this study for you?
When we started the project, we had the idea that we quite rapidly would find a T cell response to neoantigens encoded by the FLT3D835Y mutation. However, after negatively screening 15 donors, we got a bit gloomy. We were successful only with donor 16, indicating that this neoantigen is not very immunogenic. This shows that T cells in patients indeed might need help to efficiently recognize this, and probably many other recurrent driver mutations. The other surprise was that T cells expressing the FLT3D835Y TCR indeed could eliminate all the patient-derived leukemia cells with the capacity to propagate leukemia in mice. We were so excited that our TCR could mediate killing of cells with the characteristics of leukemia stem cells!
What is the outlook?
We would of course like to see our TCR in the clinic. Translating new therapeutic concepts in cell therapy is, however, challenging in academia, both from a logistic and resource perspective. I believe that the government has a responsibility to ensure that promising novel concepts are tested in phase I trials. The pharmaceutical industry is risk-averse, and wants to see proofs-of-concept in humans before they commit. If only projects with the potential to reach very large patient groups and become blockbusters are funded, many smaller patient groups might never get curative therapies.
What was the coolest thing you’ve learned (about) recently outside of work?
I have three boys, and my teenage son is making rap music in his spare time together with his buddies. Learning about how they build up a song, sequentially adding layers, has opened a new world to me, quite different from when I used to play the piano as a kid...
Otherwise, during a busy day at work and at home, luxury for me is to be able to run home from work. After taking the bus four stops from the hospital, I run along a river with waterfalls, bird songs, squirrels, deer, and beavers, not seeing a single car and ending in my backyard. Keeps me sane!



