This week’s extensive special feature covers select talks from the AACR Tumor Immunology and Immunotherapy Meeting 2023 in Toronto, Canada. We have organized the content by topics below.
Keynote lecture
Crystal Mackall
T cell therapies
Phil Greenberg
Carl June
Greg Delgoffe
Cytokine therapies
Pablo Umana
Ivana Djuretic
David Brooks
Tumor immune microenvironment
Catherine J. Wu
Thomas Gajewski
Paul Thomas
Robert Schreiber
Immune regulation
Matthew Krummel
Ping-Chih Ho
Lloyd Bod
Brian Brown
Keynote lecture
Chasing T cell Exhaustion - Crystal L. Mackall - Stanford University School of Medicine, Stanford, California
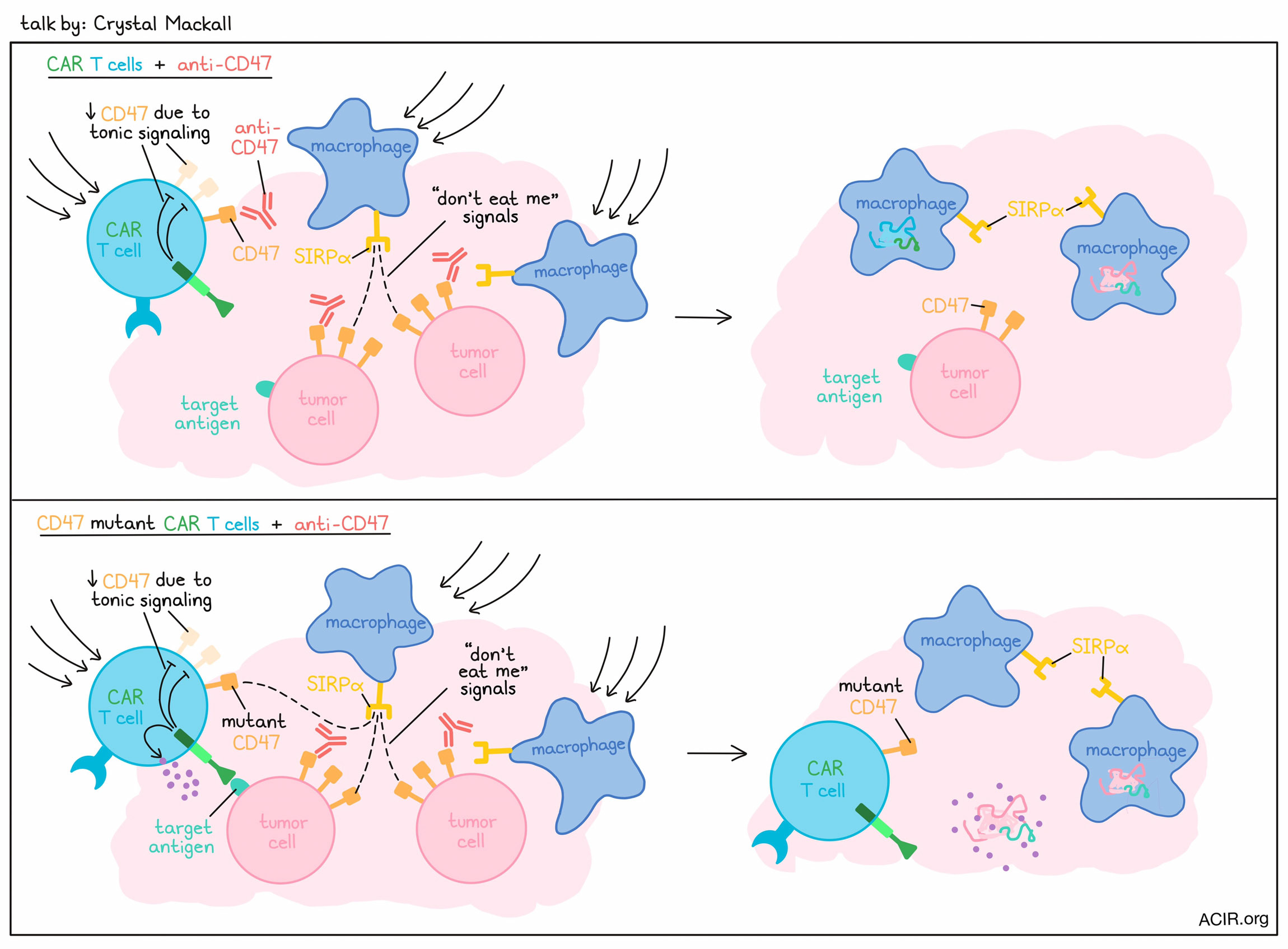
In the opening keynote lecture, Crystal Mackall described how cancer immunotherapy may not always benefit most from maxing out responses, but rather, by tuning responses to an optimal level. First, Mackall described studies in which GD2.28.z CAR T cells failed in osteosarcoma models due to early exhaustion induced by tonic signaling associated with scFv aggregating on itself in membranes. This could be remedied by switching out the CD28 costimulatory domain for a 4-1BB costimulatory domain. While the 4-1BB domain is not as strong, it allowed for reduced tonic signaling and exhaustion, endowing the CAR T cells with enhanced antitumor efficacy. Further investigating the potential to mitigate exhaustion in T cells, Mackall described a project in which exhaustion was found to be regulated by a balance of transcription factors that either promoted or prevented exhaustion programming. Altering the balance of these transcription factors through knockout or forced overexpression could effectively control exhaustion. In particular, overexpression of c-Jun was found to prevent exhaustion indirectly by preventing the formation of inhibitory transcription factor heterodimers via the bZIP domain. While this system induced exhaustion programming through changes in transcription factor expression, it did not alter the epigenome. Her group also found that dasatinib could transiently and reversibly inhibit CAR T cell signaling, preventing proliferation, cytokine secretion, and cytotoxicity through inhibition of Lck. Unlike in the previous project, this strategy of “resting” CAR T cells did alter the epigenome, opening and closing genes that suppressed and promoted exhaustion, respectively. The team noted a group of genes that were uniquely expressed in this rested state, including TCF7 and FOXO1. Investigating in mice, the researchers found that knockout of FOXO1 drove exhaustion programming, while overexpression of FOXO1 drove stemness programming and enhanced potency. Interestingly, altering FOXO1 expression also drove epigenetic changes, while altering TCF7 expression did not, suggesting that TCF7 may be more of a marker of FOXO1 activity than a driver of stemness. Discussing why FOXO1 may not have been identified previously, Mackall explained that it is not regulated by RNA levels, but rather, by nuclear shuttling. Based on this, the team found that the genes altered by FOXO1, the “FOXO1 regulon”, correlated with CAR T and TIL responses and could identify potent T cells. Mackall also described novel SNIP-CARs engineered with a cut site for the NS3 protease. Under standard conditions, the protease would cleave the CAR and prevent signaling, but the addition of a drug could inhibit the protease and allow for the CAR to remain intact and exert its signaling functions. This system allows for an effective “on” switch that was intended to enhance safety, but interestingly, it also enhanced stem-like and effector phenotypes and improved the antitumor efficacy of CAR T cells. This was found to be attributed to the daily dosing of the drug, which led to oscillation of drug levels that allowed the CAR T cells to “rest” between doses. Switching the conversation from T cells to macrophages, Mackall discussed research in which blockade of CD47 – the “don’t eat me’ signal” – abrogated CAR T cell activity, dependent on the presence of macrophages, but not related to ADCC. Meanwhile, CD47 overexpression on CARs allowed the CAR T cells to persist, improving antitumor efficacy. In co-culture experiments, macrophages were found to preferentially phagocytose activated CAR T cells, even in the absence of anti-CD47. Based on the known role of CD47 as a signal that is lost in aging blood cells, the researchers hypothesized that it may play a similar role in T cells. Investigating, they found that tonic CAR signaling drove reduced CD47, increasing susceptibility to phagocytosis. This phenomenon was also evident in a patient, where CAR mRNA could be identified in the myeloid compartment, consistent with phagocytosis of engineered cells. To prevent macrophages from eating activated CAR T cells and to simultaneously take advantage of phagocytic macrophages against tumors, Mackall and colleagues developed a mutant CD47 that would bind SIRPα, but not anti-CD47. When CAR T cells were engineered to express this mutant CD47, they became resistant to anti-CD47/macrophage-mediated depletion, and instead, the macrophages drawn in by the CAR T cells showed activity against tumors. This synergistic activity between the mCD47 CAR and the anti-CD47 demonstrated the value of tuning responses in macrophages, which may represent a fundamentally common pathway for maintaining T cell homeostasis for highly activated T cells.
T cell therapies
“Ain’t no mountain high enough”: Making TCR-T cells work against solid tumors - Philip D. Greenberg - Fred Hutchinson Cancer Research Center, Seattle, Washington
Phil Greenberg discussed the use of TCR-engineered T cells against solid tumors. In order to isolate high-affinity native TCRs for defined antigen targets, Greenberg and colleagues developed a rapid high-throughput method involving the stimulation of healthy donor PBMCs with autologous peptide-loaded DCs and cytokines, followed by high-stringency sorting of antigen-specific CD8+ T cells. Using this strategy, they were able to identify and engineer TCRs for the mesothelin antigen present in murine pancreatic cancer (PDA in B6 KPC mice). Upon transfer, TCR-engineered T cells preferentially accumulated in spleens and tumors, killing tumor cells, dissolving much of the tumor structure, opening up blood vessels, and relieving the dense pressure associated with fibrosis, allowing patency. Over time, T cells increased a TGFβ response gene signature, expressed more inhibitory receptors, and became more homogeneous and dysfunctional, though T cell activity could be sustained for longer with repeated infusions. Based on this preclinical evidence, the researchers organized a clinical trial in which patients with metastatic PDA were treated with 3 doses of mesothelin-specific TCR-engineered T cells. Thus far, dose levels 1 and 2 have been well tolerated, and the first patient at dose level 3 – the first dose at which the researchers might expect to see results – was very recently treated. Engineered cells detected in tumor biopsies from these early patients show acquisition of TGFβ response and dysfunctional gene signatures by day 21, matching the mouse model. Going back to their mouse models in search of targets to modulate, the researchers used scRNA and ATACseq co-assays to parse out distinct chromatin landscapes in different TIL populations, and subsequently identified 2 specific transcription factors, SMAD3 and FLI1, that bound to different clusters of regulatory regions, and may play a role in dysfunction. The team also used an in vivo CRISPR screen to target 200 genes based on their upregulation or downregulation in clusters from infiltrating cells from the KPC mouse model. In their early data, they are already seeing several expected patterns, and hope that further investigation will provide more insight. Next, Greenberg discussed the possibility of using next-generation synthetic biology strategies to overcome obstacles like poor T cell survival and dysfunction. One such strategy is using immunomodulatory fusion proteins (IFPs) that can convert inhibitory/death signals to costimulatory, proliferation, and survival signals by replacing the inhibitory endodomains on molecules like PD-1, CD200R, and SIRPα with the endodomains of costimulators like CD28 or 4-1BB. Another strategy, Greenberg described, is to insert a CD8+ T cell-derived TCR targeting a known class-I-restricted antigen (WT1) into CD4+ T cells together with the CD8 co-receptor to enhance direct antitumor activity and enhance help for WT1-specific CD8+ T cells. Greenberg also applied this strategy to an engineered TCR specific for a shared KRAS mutation. With promising preclinical results, an IND was recently approved for a trial in PDA, NSCLC, and colorectal cancer with CD4+ and CD8+ T cells expressing the engineered TCR and CD8αβ. Furthermore, addition of FAS-4-1BB has been shown to enhance serial killing of tumor cells by the engineered T cells in preclinical studies, so an IND has been filed for the evaluation of TCR+CD8αβ+FAS-4-1BB+ CD4+ and CD8+ T cells in a follow-on trial. One challenge to the clinical translation of fusion proteins/switch receptors for TCR gene therapy is that the size of multicistronic vectors gets quite large. To shorten the TCR plus CD8αβ plus fusion protein construct, the researchers instead tethered the CD28 costimulatory domain to the CD8αβ molecule, adding only 84 base pairs, compared to the ~700 base pairs that the addition of an extra immunomodulatory fusion protein would add. Expression of the chimeric CD8–CD28 receptor reduced dysfunction and promoted more proliferation, tumor infiltration, and tumor killing. Finally, Greenberg described a strategy to address immunosuppressive cytokines in the TME using TGFβR chains redesigned to deliver an IL-2 signal, converting an otherwise inhibitory signal into a costimulatory signal. These various strategies show strong promise for the use of TCR-engineered T cells and IFPs to tackle many of immunotherapy’s biggest hurdles.
Gene deletions unveiled: Cracking the code for CAR T cell supremacy - Carl H. June - Perelman School of Medicine University of Pennsylvania, Philadelphia, Pennsylvania
Discussing CAR T cells,Carl June described both the impressive responses in patients with ALL and the less impressive responses in those with CLL, noting that there is likely an issue with T cell dysfunction in these patients. In an effort to reduce CAR T cell dysfunction, June and colleagues recently tested the possibility of knocking out the inhibitory checkpoint receptors CTLA-4 and/or PD-1. While they hypothesized that double knockout would be superior, they instead were surprised to find that while single knockout of CTLA-4 improved the maintenance of CAR expression and enhanced the effector functions and antitumor efficacy of CAR T cells under conditions of long-term exposure to tumor cells/antigens, PD-1 single knockout had no effect. Further, in double-knockout cells, PD-1 knockout even abrogated the desirable effects of CTLA-4 knockout. The effect of CTLA-4 knockout was found to be due to enhanced CD28 signaling, which supported cytokine production and more central memory-like phenotypes. This effect was nullified in cells that were also PD-1-deficient. In mice, CTLA-4 KO, but not PD-1 or double KO CAR T cells resisted exhaustion and mediated enhanced antitumor immunity and longer survival in mice with ALL or MM, regardless of whether the CAR T cells were derived from healthy donors or patients with CLL. Further, CTLA-4 is often upregulated on CAR T cells in patients who are non-responders to therapy, suggesting clinical relevance.
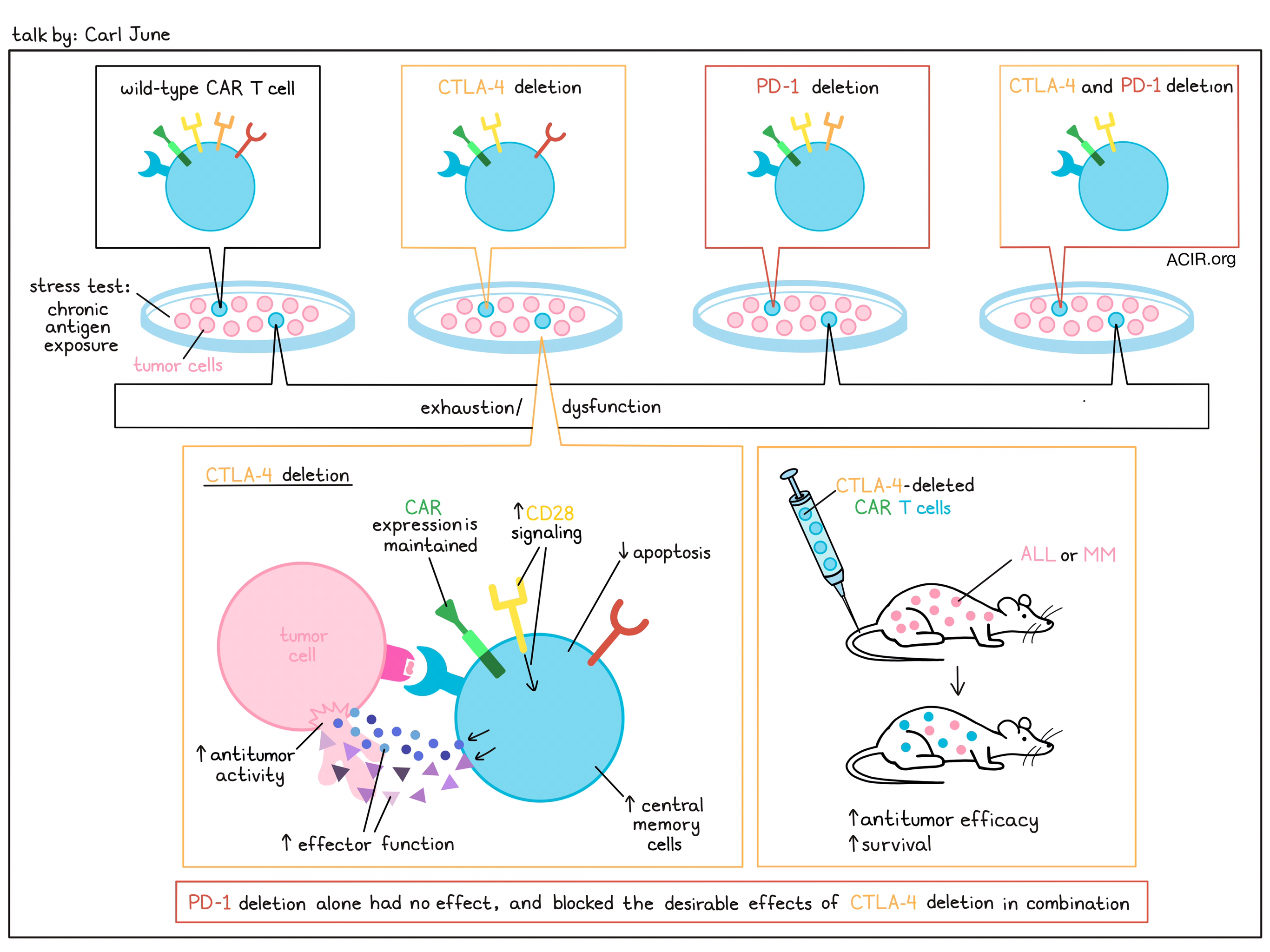
In another study, June and colleagues evaluated the possibility of editing out multiple genes across multiple chromosomes using CRISPR/Cas9. While this was shown to be feasible, some chromosomal translocations were detected, and CRISPR/Cas9 editing at the TRAC locus (to knock out the native TCR) resulted in chromosome loss, as did homology-directed repair. To mitigate this issue, June’s team developed a clinical CRISPR/Cas9 editing process that optimized the cell manufacturing process to preserve chromosomal integrity, which required p53 expression. Finally, June described experiments involving the knockout of Tet2. These showed that when CARs were integrated into the TET2 locus, CAR T cells performed better. TET2 loss was found to induce chromatin accessibility at TOX and TOX2 sites. While these TOX family molecules are often associated with exhaustion, the role of TOX2 in human T cells is not well documented. In recent work, June and team showed that TOX2 regulates central memory differentiation in human CAR T cells, while TOX was required for effector memory T cell differentiation. These effects were more pronounced in CD4+ versus CD8+ T cells. Interestingly, while TOX expression supported TOX2 expression in mice, it appeared to downregulate TOX2 in human T cells, suggesting different roles in the different species, and a non-redundant role for TOX2 as a potentiator of central memory differentiation in human T cells.
Metabolic modulation during in vitro expansion for curative adoptive cell therapy for cancer - Greg M. Delgoffe - UPMC Hillman Cancer Center, Pittsburgh, Pennsylvania
CAR T cells come in many shapes and sizes, and have shown impressive results in patients, but still many CAR T cell treatments fail, often due to lack of persistence. Addressing this issue, Greg Delgoffe and colleagues investigated ways to modulate CAR T cell metabolism, particularly during the ex vivo expansion process. Looking at metabolism in naive and memory T cells, which are known for quiescence and long-term persistence, the researchers found that memory T cells have high mitochondrial mass, allowing for a high respiratory capacity that can be quickly deployed upon activation/reactivation. Tumor-infiltrating lymphocytes, on the other hand, tend to quickly acquire stress-related metabolic deficiencies, including reduced glucose uptake capacity and losses in functional mitochondria. This reduction in mitochondria is mediated by repression of PGC1a – a transcriptional activator of mitochondrial biogenesis. Based on this evidence, Delgoffe and colleagues overexpressed PGC1a in TCR-engineered or CAR T cells, and found that it dramatically improved T cell responses and outcomes. Looking at how current ex vivo expansion conditions (high IL-2, readily available glucose and metabolites, and hyperoxygenation) affect CAR T cell metabolism, Delgoffe’s team found that CAR T cells become hypermetabolic. The researchers therefore hypothesized that while this T cell expansion method may enhance activation, it may also limit persistence. To test this, they compared ex vivo activation (with SIINFEKL peptide and APCs) and in vivo expansion (with vaccination with Vaccinia expressing OVA) of OT-1 cells and saw strong divergence at the metabolic level, with T cells expanded in vivo showing higher mitochondrial mass, lipid storage, and respiratory capacity. While there was no difference in the ability of these cells to take up glucose, they utilized glucose differently, with ex vivo-expanded cells performing less aerobic glycolysis. Investigating what drives this, Delgoffe noted their previous work, which showed that PDH, a gatekeeper enzyme of glucose metabolism, can be inhibited by an inhibitory kinase (PDKH1), which is activated during T cell activation. Inhibition of PDH diverts pyruvate to exported lactate rather than importing it into mitochondria to feed the TCA cycle during aerobic glycolysis. By targeting PDHK1 with dichloroacetate (DCA), glycolysis could be restored to baseline, reducing mitochondrial stress, increasing mitochondrial biogenesis, and restoring the respiratory capacity of therapeutic T cells. In mouse models, therapeutic T cells cultured in the presence of DCA showed superior antitumor immunity and improved survival compared to T cells cultured under the standard conditions without DCA, and were superior even without lymphodepletion prior to transfer. Similar results were observed using human T cells against tumors in xenograft mouse models. These results were not due to changes in exhaustion or effector function, or differences in mitochondrial mass, but rather, due to a numerical advantage, as DCA-expanded T cells were more prominent in both tumors and lymph nodes. This was found to be due to enhanced survival of DCA-expanded T cells, beginning shortly after infusion and lasting long-term – even after tumors were cleared, or in mice that were tumor-free to begin with. DCA-expanded T cells were also found to be more stem-like and more adept at oxidative phosphorylation in the presence of alternative carbon sources, suggesting traditional ex vivo expansion techniques may cause T cells to develop a glucose addiction, and that glucose may not actually be the optimal food source for T cells. Further, DCA promoted increased carbon flux into the nucleus, promoting the acetylation of histones around key genes for stemness and central memory. This effect was due to increased leaking of citrate (a precursor to Acetyl-CoA, which is required for histone acetylation) from the mitochondria, and thus increased mitochondrial crosstalk with the nucleus. These results underscore the importance of metabolism in T cell therapies, and suggest that protocols may benefit from modifying the flux of glucose and other carbon sources to sustain respiration and promote T cell survival and persistence in patients.
Cytokine therapies
PD1-IL2v: PD-1-Cis IL-2Rβγ agonism yields better T cell effectors from stem-like CD8+ T cells - Pablo Umana - Roche, Schlieren, Switzerland
Pablo Umana and colleagues synthetically engineered a target-guided IL-2 mutein suitable for systemic cancer therapy. Although IL-2 is a potent cytokine for cancer immunotherapy, only a subset of patients (15%) benefit from high-dose IL-2 therapy, and its clinical use is limited by its severe side effects (such as capillary leak syndrome), short half-life, poor tumor targeting, and Treg expansion. The differential expression of the IL-2 receptor chains (IL-2Rα, β, and γ) on immune and endothelial cells controls the affinity with which IL-2 is bound by these cells, and dictates the effects of systemic IL-2 administration. Endothelial cells, Tregs, and Teff cells express the high-affinity IL-2 receptor, (IL-2Rαβγ) on their cell surface, while naive T cells, CD8+ memory T cells, and NK cells, express the intermediate affinity IL-2 receptor, IL-2Rβγ. Umana and colleagues mutated IL-2 (IL-2v) to abolish IL2Rα binding, and attached it to an antibody targeting the tumor stroma antigen FAP. The resulting immunocytokine, FAP–IL2v, demonstrated improved tolerability compared to high-dose IL-2 therapies, expanded total CD8+ T and NK cells, but not Tregs, and induced responses in some patients who had progressed on checkpoint blockade. In a chronic LCMV model, only the combination of anti-PD-L1 with FAP–IL2wt, but not FAP–IL2v enhanced the expansion of antigen-specific CD8+ T cells. To increase the activity of the IL-2v on antigen-specific effector cells, the researchers fused the IL-2v with a high-affinity anti-PD-1 antibody (PD1–IL2v). PD1-IL2v targeted PD-1+ T cells, bypassed Tregs, and fully retained activity on NK and CD8+ T cells via IL-2Rβγ activation. PD-1-mediated IL-2v delivery resulted in enhanced IL-2Rβγ activation. In an orthotopic pancreatic cancer model (PancO2) PD1-IL2v demonstrated superior efficacy compared to FAP–IL2v, and in a subcutaneous PancO2 model, PD1–IL2v induced polyfunctional CD8+ TILs and increased the CD8/CD4 T cell ratio in tumors compared to blood. Back in the chronic LCMV model, cis-targeted PD1–IL2v increased the expansion of antigen-specific and polyfunctional CD8+ T cells, and yielded better effectors from stem-like precursors with reduced terminal exhaustion markers compared to trans-targeted FAP–IL2v in combination with anti-PD-L1. PD1–IL2v generated effector T cells with unique epigenetic and transcriptional profiles that were more cytotoxic and less exhausted. In a B16-OVA model, PD1–IL2v showed superior tumor eradication over FAP–IL2v plus anti-PD-1 and enhanced efficacy in combination with anti-PD-L1 in the autochthonous Rip-Tag5 pancreatic neuroendocrine cancer model. In summary, shifting IL-2Rβγ agonism from CD25 to PD-1 cis-targeting differentiates precursor exhausted CD8+ T cells into better effectors.
Restricting the activity of IL-2 and IL-21 to CD8+ T cells greatly enhances their therapeutic potential - Ivana Djuretic - Asher Biotherapeutics, Inc., South San Francisco, California
Cytokines are powerful immunotherapies, but are also pleiotropic and often induce both desired and undesired consequences, limiting their activity and driving toxicity. Ivana Djuretic and colleagues used cis-targeting to deliver a selected cytokine only to targeted cell types expressing the cytokine receptor. For cis-targeting, high-affinity cytokines are attenuated to decrease the affinity of the cytokine to its receptors and reduce the signaling in all cell types, and then fused to an antibody that recognizes an antigen on the target cell to increase its local concentration, thereby increasing selectivity. IL-2 that doesn’t bind to its alpha receptor (Not-a IL-2) presented limited toxicities and modest activity with preferential NK cell expansion in clinical trials. To test the therapeutic index of Not-a IL-2 in mice, the researchers treated MC38 mouse tumors with Not-a IL-2 and observed higher toxicity at doses with antitumor activity and preferential NK cell expansion, similar to human trials. In the MC38 tumor model, CD4+ T cell, CD8+ T cell, or NK cell depletion showed that CD8+ T cells drove the antitumor activity, while NK cells were responsible for the toxicity. Ivana Djuretic and colleagues engineered AB248, a CD8b-targeted IL-2 mutein fusion molecule (a Nota-IL-2 with reduced βγ receptor binding) that preferentially activates CD8+ T cells with over 500-fold selectivity compared to NK and Tregs. Selective CD8+ T cell targeting decreased the NK cell-dependent cytokine release syndrome, including IFNγ production in human peripheral blood mononuclear cells. In an MC38 tumor model, mouse CD8-mIL2 preferentially expanded CD8+ T cells in blood and tumors, and showed strong monotherapy antitumor response (7/10 CR) with a single dose, without body weight loss. In an anti-PD-1-resistant T3 sarcoma model, CD8-mIL2 synergized with anti-PD-1 therapy, leading to 5/5 CR and expansion of tumor antigen-specific CD8+ T cells. Immunophenotyping of antigen-specific CD8+ T cells showed increased expression of granzyme B and decrease in TOX and PD1+, TIM3+ CD8+ T cells. Single-cell RNAseq further showed that CD8-mIL2 expanded CD8+ T cell clusters with effector and memory phenotypes. In non-human primates, AB248 was well tolerated, with no evidence of vascular leak or other IL-2-related toxicities, and showed selective CD8+ T cell expansion at a relevant dose. Ivana Djuretic then introduced a second immunocytokine, AB821, a CD8+ T cell-selective IL-21 mutein (reduced IL-21R binding) with over 1000-fold in vitro selectivity for CD8+ T cells and robust monotherapy activity. CD8-mIL21 showed high CR rates as a monotherapy in select PD-1-resistant models, and synergized with anti-PD-1 in resistant (Pan02 and B16F10) models. Single-cell RNAseq analysis of CD8+ T cells showed remodeling of the CD8+ T cell compartment from majority exhausted clusters to granzyme B- and perforin-expressing functional clusters, and strong clonal expansion with CD8-mIL21, distinct from CD8-mIL2. IL-2- and IL-21-driven enhancement of CD8+ T cell effector functions were TCR-dependent.
Interferon modification of T cell function and response to immunotherapy - David G. Brooks - University of Toronto, Toronto, Ontario, Canada
David Brooks emphasized on the underlying role of type 1 interferon (IFN-I; in particular IFNα/β) signaling in T cell function and response to immunotherapy. Multiple inflammatory and immunosuppressive mechanisms inhibit immunity during viral or tumor persistence, such as alterations in innate immunity, CD4+ Th cell skewing, B cell dysfunction, CD8+ T cell exhaustion, and infected or tumor cell-intrinsic immune resistance. While IFN-I signaling is critical for viral control and to stimulate the immune response to viral infection, chronic IFN-I signaling induces immunosuppression, maintaining viral persistence. Transient blockade of IFN-I signaling at a very early stage (day -1 to day 8 post infection) enhanced control of chronic LCMV infection. Likewise in cancer, IFN-Is are important at all stages of disease progression and are critical for beneficial response to radiation and immunotherapy. In contrast, IFNs also drive therapy resistance to radiation and immunotherapies. To understand what determines the opposing outcomes of IFN-I signaling, Brooks and team focused on the role of interferon regulatory factor 2 (IRF2), a repressor of IFN-related gene expression. IRF2 is broadly expressed in various tumor-infiltrating immune cell types in humans and mice. IRF2 knockout mice engrafted with IRF2-sufficient tumor cells (MC38, B16, or PyMT) were able to control tumor growth, demonstrating that IRF2 deletion in immune cells enabled control of diverse tumor types. To determine which immune cells were associated with the response, the researchers deleted IRF2 specifically in CD8+ T cells and saw similar tumor control. Consistently, adoptive transfer of IRF2-deficient tumor-specific CD8+ T cells delayed the growth of established tumors. IRF2-/- tumor-infiltrating CD8+ T cells expressed intermediate levels of exhaustion markers, were able to induced high levels of cytokines, granzyme B, and perforins, and were enriched for an effector gene signature, but did not show significant epigenetic changes. Furthermore, the transcriptional profile of IRF2-/- tumor-infiltrating CD8+ T cells revealed decoupling of enhanced IFN signaling from an exhaustion phenotype. Blockade of either type I or type II interferons (IFNγ), individually or together, led to loss of tumor control, signifying that enhanced IFN-I and IFNγ signaling are critical for tumor control in IRF2-deficient mice. To understand the complexity and diversity of human IFN-I responses to immunotherapy, Brooks and colleagues focused on the single-cell expression patterns of IFN-I reactivity. Baseline patient PBMCs prior to treatment stimulated with IFNβ induced interferon-stimulated proteins (ISPs). Average fold-changes in the expression of 7 ISPs were used to determine IFN-I responsiveness capacity scores (IRCs). Low CD4+ and CD8+ T effector cell responsiveness to IFN-I stimulation in PBMCs taken at the baseline prior to anti-PD-1 blockade in patients with HNSCC or melanoma correlated with increased progression-free and overall survival following anti-PD-1 blockade. The researchers then incorporated CD4+ Teff IRC, CD8+ Teff IRC and myeloid cell IDO induction, and found that patients who were predicted to respond to therapy based on their baseline IFN-I sensitivity indeed showed greater long-term overall survival to anti-PD-1 therapy. Mechanistically, differences in IFN-I responsiveness were determined by the physiological cell state. Patients with high IFN-I responsiveness had open chromatin regions enriched in IFN-inducing transcription factors and were poised to react to IFN-I-induced transcription factors.
Tumor immune microenvironment
T cell specificity, phenotype, and dynamics: Impact on cancer immunotherapy - Catherine J. Wu - Dana-Farber Cancer Institute, Boston, Massachusetts
Catherine Wu gave an overview on the native landscape of tumor-reactive tissue-infiltrating T cells. TCRs with antitumor specificity in the tumor immune microenvironment are rare at baseline, which is dominated by bystander cells. The researchers hence focused on the single-cell characterization of TILs from patient’s tumors to understand the cellular phenotypes and TCR clonotypes; the generation of patient-derived melanoma cell lines to determine tumor-specificity; and the reconstruction and expression of TCRs in vitro to screen TCR antigen specificities. To screen TCRs and antigens in higher throughput, the group developed a medium throughput strategy combining TCR cloning and expression in donor T cells combined with combinatorial dye labeling of each clonotype-bearing cell so that tens of TCRs can be interrogated in one tube against tumor or antigen targets. Deep characterization of CD8+ T cells in patients with melanoma showed exhausted T cell and non-exhausted memory T cell clusters. Further analysis showed that the dominant TCR clonotype clonally expanded in the exhausted T cell population had 83% of tumor-specific clonotype either against melanoma-associated antigens or neoantigens whereas the dominant TCR clonotype in the non-exhausted memory T cell compartment had 10% tumor-specific and 11% EBV- or virus-specific clonotypes. Similarly, in CD4+ T cells, the dominant TCR clonotype in exhausted and Treg compartments had 84% tumor-specific TCRs, whereas memory T cells had 5% tumor-specific TCRs. Antigen specificity across 4 patients showed different antigen profiles, with two of the patients enriched for melanoma-associated antigens, and the other two with neoantigens and viral antigens, suggesting tumor-intrinsic features drive the antigen-specific immune response and TIL landscape. A putative tumor-specific CD8+ T cell signature derived from the melanoma single-cell transcriptome held in RCC and HNSCC. In another study, patients with stage III/IV HPV-unrelated HNSCC treated with 2 rounds of neoadjuvant anti-PD-1 showed evidence of pathological response. Phenotypic characterization of TILs in responders and non-responders showed responders were enriched in exhausted T cells with high cytotoxic and tissue-resident programs. Comparison of pre- and post-treatment TILs suggested that response was predetermined and associated with the contraction of Tex with a TRM program in responders. Mechanistically, clonal replacement and revival were not involved, but tumor-reactive T cells with high ZNF683 expression and high cytotoxic potential were present at baseline. Contraction of expanded ZNF683high T cells expressing a signature of cytotoxicity was associated with response to therapy, and the contraction was most likely related to antigen clearance. Wu dubbed this novel process “cytotoxic revival”. (We are also grateful to Cathy for giving a shout-out for ACIR’s coverage of this work and our mission in general!) Citing parallel work from her lab in other disease settings, Wu described how in patients with Richter syndrome, ZNF683 regulated pathways related to activation and cytotoxicity; ZNF683high T cell infiltration was associated with better outcomes in solid tumors; and ZNF683high signatures and ZNF683-expressing T cells could be detected in the blood and were associated with response to anti-PD-1 therapy. Similarly, ZNF683high T cells were expanded in PBMCs in AML responders compared to non-responders following transplant and donor lymphocyte infusion. Finally, Wu transitioned to her group's work in vaccination. In melanoma patients, vaccination generated memory response, T cell diversification and persistence, recruitment of T cells to tumor sites, and epitope spreading. In patients with high-risk ccRCC, neoantigen-targeted personalized vaccines in the adjuvant setting, with or without ipilimumab administered locally to the vaccine site, led to recurrence-free survival in all patients up to 40 months. Vaccine-associated immune responses persisted a year after vaccine, and ipilimumab dramatically increased the diversification of T cell clonotypes.
Critical role for Batf3-lineage dendritic cells within the tumor microenvironment - Thomas F. Gajewski - University of Chicago, Chicago, Illinois
Thomas Gajewski presented some exciting work on the role of Batf3-lineage dendritic cells in optimal CD8+ T cell priming in the draining LN and the recruitment and activation of effector CD8+ T cells back to and in the TME. T cell-inflamed and non-inflamed tumor microenvironments escape immune surveillance via distinct mechanisms. Inflamed tumors have higher cytokines, CD8+ T cell infiltrations, increases in type I IFN signatures, and escape immunity via upregulation of inhibitory pathways, while non-inflamed tumors have low inflammatory signatures and lack CD8+ T cells, escaping immunity via T cell exclusion. Several factors, such as tumor cell-intrinsic somatic mutations, the composition of commensal microbiota, and host germline genetic differences are some major sources of inter-patient heterogeneity that regulate T cell-inflamed tumor microenvironment. With respect to tumor cell-intrinsic genetics, WNT/b-catenin activation prevents Batf3 DC recruitment, causing resistance to checkpoint blockade, vaccination, and adoptive T cell therapy. In a Braf/PTEN GEMM melanoma model, loss of Batf3 DCs prevents recruitment of effector CD8+ T cells, which is rescued by Flt3L-activated DC injection, suggesting that CD8+ T cell recruitment in the tumor is Batf3 DC-dependent. In human melanoma, transcripts for CXCL10, a major T cell-recruiting chemokine produced by cDCs, are associated with a T cell-inflamed phenotype and clinical response. Batf3 DC, but not antigen presentation, is the rate-limiting step in non-inflamed tumors. To test if Batf3 DCs are required during PD-1/PD-L1 blockade to provide signals for TIL reinvigoration within the TME, Gajewski and colleagues selectively depleted DCs within the tumor microenvironment immediately prior to PD-L1 blockade, and found that anti-PD-L1 efficacy is lost upon selective DC depletion. Based on the high expression of 4-1BBL on DCs, they tested manipulating this activating pathway. 4-1BB blockade abolished the anti-PD-L1 efficacy, whereas direct agonistic anti-4-1BB stimulation of T cells rescued anti-PD-L1 efficacy in absence of host DCs, signifying that anti-PD-L1 efficacy is dependent on host 4-1BBL. In anti-PD-1-treated melanoma patients, spatial analysis of CD8+ T cells and Batf3+ DCs showed non-random distribution associated with anti-PD-1 efficacy, and the close proximity of CD8+ T cells to cDC1s within the TME was associated with multiple signals of effective T cells. Gajewski then turned to how to solicit Batf3 DCs to establish a T cell-inflamed TME and enhance checkpoint blockade efficacy. Intratumoral injection of modified STING agonists trigger durable rejection in CT26 and 4T1 tumor models, but these transplantable tumors have some degree of spontaneous inflammation and are not cold tumors. The B-Raf/PTEN-/-b-catenin (BPC) GEMM model has a cold TME and doesn’t respond to checkpoint inhibitors. STING agonist (DMXAA) failed to control tumors in BPC GEMM as monotherapy, and in combination with ICBs. DMXAA did induce T cell infiltration, but failed to increase the CD103+ DC infiltration in BPC tumors needed to activate these cells. To test if Flt3 ligand can increase CD103+ DC infiltration in non-inflamed tumors, Gajewski and colleagues treated BPC tumors with Flt3L followed by DMXAA. The combination improved tumor control in mice subsequently treated with anti-PD-L1 and anti-CTLA-4.
Specificity matters: The breadth of endogenous and elicited antitumor T cell responses - Paul G. Thomas, St. Jude Children’s Research Hospital, Memphis, Tennessee
Paul G Thomas presented his work on TCR features associated with antiviral and antitumor T cells responses. Paul introduced TCRdist, an algorithmic tool that identifies core motifs of specificity by comparing the sequences across relevant regions of TCR recognition (with over-weighting of CDR3) between two TCRs, and scores the similarities and differences in a structurally informed way. However, similarity matching alone is not enough to determine the TCR antigen recognition without defining the likelihood of TCR–antigen interaction, and importantly, additional tools are needed to correct for the generative probability of TCRs. Analysis of relatedness scores across a large population of cells can identify clusters of sequence-related clonotypes, which is typical of viral infection (“viral clusters”). The researchers then used these tools to characterize the TCR repertoire in COVID19 patients who received two doses of the BioNTech/Pfizer vaccine. Bulk TCR sequencing data was used to identify the specificity clusters, which showed clusters of closely related TCRs, with a particularly dramatic clustering in one TCR beta chain, suggesting immunodominance. Using a MIRA dataset, the researchers mapped the single chain of the immunodominant clone to the specific region of the spike protein of the virus and the individual HLA-peptide MHC associated with it. The repertoire was stable for at least six months. Longitudinal analysis of acute versus convalescent blood samples from COVID19 patients showed clusters of highly similar TCR sequences in blood associated with COVID19 response. In an influenza virus mouse model, more than 60% of the response is against a single epitope and knockout of the dominant epitope changed the response towards other epitopes, supporting immunodominance and suggesting that immunodominant epitopes can be changed. Thomas then presented his work on pediatric cancers, which often have fusion-driven mutations (which, based on their work, are highly immunogenic) and a low tumor mutational burdens; hence, the responding repertoires may be limited by immunodominance. In acute lymphoblastic leukemia, every patient had a conserved ETV6-RUNX1 fusion antigen as a public neoantigen. Although immunodominance limits the responding repertoires in patients with high tumor mutation rates, and thus a high likelihood of truly immunodominant epitopes, about 86% of the mutations in these patients were targeted by T cell responses in the tumor at the time of diagnosis. These cells appeared largely non-exhausted, however, and pediatric cancers are typically unresponsive to ICB, suggesting other limitations in the effectiveness of these T cells. In another study in patients with AML and MPAL, tumor-specific T cell responses could be identified in nearly all patients, except those with HLA downregulation. TCR cloning showed the presence of the immunodominant fusion epitope KMT2A::AFF1, but clones from a patient showed only a single TCR clonotype with no related clonotypes. In Fibrolamellar carcinoma, which is driven by DNAJB1-PRKACA gene fusion, ex vivo-expanded TILs responded to the DNAJB1-PRKACA neoepitope peptide. A reactive clonotype was quickly identified, but after searching thousands of clonotypes found in the blood and tumor of the patient, only one additional clonotype could be found; only the original TCR was effective as a TCR therapy. In an ongoing 16-patient clinical trial of a Fibrolamellar hepatocellular carcinoma, a DNAJB1-PRKACA fusion kinase peptide vaccine combined with nivolumab and ipilimumab led to 2 complete responses. In patient 5 (a CR), top responding clones are mostly all CD4+ with a Th17-like phenotype, and TCRdist revealed multiple “viral clusters”.
Hyperactivated CD4+ T cells lacking FoxP3 inhibit therapeutics tumor immunity - Robert D. Schreiber - Washington University School of Medicine in St. Louis, Missouri
Building on a history of research into neoantigen vaccines, Bob Schreiber discussed recent and ongoing work on the contributions of MHC-I and MHC-II neoantigens in personalized cancer vaccines. Using their 129S6 Strain T3 MCA sarcoma model, which has well defined MHC-I (mLAMA4) and MHC-II (mITBG1) neoantigens, Schreiber and colleagues showed that when MHC-II neoantigens were left constant (at 1.5 ng) during vaccination, increasing doses of MHC-I neoantigens induced stronger tumor rejections that maxed out at around 150 ng. However, when MHC-I neoantigens were constant (at 150 ng) and MHC-II neoantigens were given at increasing doses, the researchers noticed more of a bell curve, where tumor rejection increased with increasing doses up to 1.5ng, then tapered off at higher doses. Investigating this observation further, Schreiber defined this lower (1.5ng), more effective dose as a low-dose vaccine (LDVax) and a higher (15µg), less effective dose as a high-dose vaccine (HDVax). Interestingly, HDVax inhibited the antitumor efficacy of anti-PD-1 (but not anti-CTLA-4) checkpoint blockade and of LDVax therapies in various tumor models. This occurred in a tumor antigen-specific manner, based on the epitopes presented on MHC-II in the vaccine and expressed in the tumors. Further, HDVax induced CD4+ T cells with markedly reduced capacity to produce IL-2, IFNγ, and TNFα, and to support CD8+ T cell expansion. Transfer of these cells into vaccine-naive T3 tumor-bearing mice inhibited the antitumor efficacy of anti-PD-1 or agonist anti-4-1BB (but not anti-CTLA-4), suggesting that these cells play an active suppressor role. While Schreiber and colleagues hypothesized that these cells were likely Tregs, scRNAseq data showed that they clustered separately from Tregs and did not express Foxp3. In a series of mouse studies, Shreiber’s team showed that both Tregs and these new suppressive CD4+ T cells independently contributed to reduced antitumor efficacy of checkpoint blockade. The researchers identified LILRB4 and Sema4a as potential antibody-targetable markers of this new cell type, and found that LILRB4 likely contributes to their suppressive functionality. Blockade of LILRB4 abrogated the HDVax-induced increases in granzyme B and CCL5 expression, and increased/normalized IL-2 production, improving tumor rejection in HDVax-treated mice. Looking at ways to overcome this HDVax-induced suppression, Schreiber’s team tested the addition of a CD8-directed IL-2 mutein, which effectively targets IL-2 to CD8+ T cells, but does not activate NK cells. This strategy restored and reversed HDVax-driven suppression, and enhanced tumor rejection. Finally, Schreiber presented data showing that while low doses of MHC-II neoantigens were presented by both cDC1s and cDC2s, higher doses were presented mainly by cDC2. Further, while the LDVax induced a predominantly CD8+ T cell responses, the HDVax induced humoral B cell responses and cytotoxic cells that actually contributed to the killing of cDC1s. Together, these results suggest that high doses of MHC-II neoantigens presented by cDC2s induce a subset of hyperactivated, cytotoxic, antigen-specific, LILRB4+ CD4+ T cells, temporarily deemed T4I, which kill antigen-expressing cDC1 and antagonize antitumor immunity. Efforts to better understand and minimize the effects of this suppressive subset could help to enhance the design of neoantigen vaccines and improve responses to immunotherapy.
Immune regulation
Immune archetypes and the shifting paradigms of myeloid biology in cancer - Matthew Krummel - University of California San Francisco Helen Diller Family Comprehensive Cancer Center, San Francisco, California
Matthew Krummel emphasized the need to look beyond the classical thinking of immune reactivity as either tolerance or destruction, and highlighted some of the emerging functions of various immune archetypes — collections of cells linked in higher-order states across tissue types. Examples include Treg/Th17 and DCs regulating commensal symbiosis, or neutrophils and macrophages mediating wound healing. As an example of tumor-immune archetypes, Krummel first showcased his group’s early work on the role of cDC1 as allies in tumor immunity, and showed that cDC1s are critical for the stimulation and proliferation of CD8+ T cells in the TME, and are associated with improved survival and response to anti-PD-L1 in mouse models. To discover dominant immune archetypes across cancers, Krummel and colleagues analyzed and sorted solid tumor samples from multiple different cancers using flow cytometry followed by RNA sequencing of different populations. Analysis of 300 patient samples showed distinct clusters of inflamed (CD8-rich) and deserted (CD8-poor) tumors. Although kidney cancer was mostly in the inflamed cluster, some of the kidney cancer samples were present in immune-desert clusters. Similar results were also found with lung cancer and melanoma, suggesting that tumor type doesn’t define the archetype. However, archetypes do link with particular immunobiology (richness of particular immune populations) and clinical outcomes. For example, in kidney cancer, patients enriched for the CD8-Mo-cDC1 archetype showed better outcomes compared to CD4-cDC2 or Treg-Mp archetypes. Krummel then presented some his lab’s work on the role of CD206+ macrophages in tumor control, demonstrating that macrophages are not actually suppressive in vivo (an in vitro non-physiologic phenotype) but rather, TAMs program T cells for exhaustion without activating them. Emphasizing the need to classify macrophages based on their function, he showed that M1 and M2 signatures were not able to classify TAMs in PyMT tumors or in human samples. In B16F10 melanoma tumors, CD206, which is considered as an M2 marker, tracks with monocyte-to-macrophage differentiation. Mechanistically, deletion of CD206+ myeloid cells in mice beginning early after tumor implantation led to indirect loss of a reactive immune archetype (decrease in MACs, cDC1, NK cells, and OT1 CD8+ T cells), but late-stage TAM depletion did not. Early depletion also decreased tumor control in B78chOVA tumors and disrupted the CXCL9:CXCR3 recruitment axis, suggesting that the CD206+ TAM:cDC1 reactive archetype is required for antitumor immunity. Comparing CD206-low and CD206-high gene sets in 230 patients from 5 indications showed a survival advantage in patients with higher CD206 expression. In summary, CD206+ TAMs in early tumor stages maintain the important CD8, NK and cDC1 archetype via the CXCL9/10 axis.
Tailoring T cell anti-tumor immunity via reprogramming mitochondria-guided transcription networks - Ping-Chih Ho - University of Lausanne, Lausanne, Switzerland
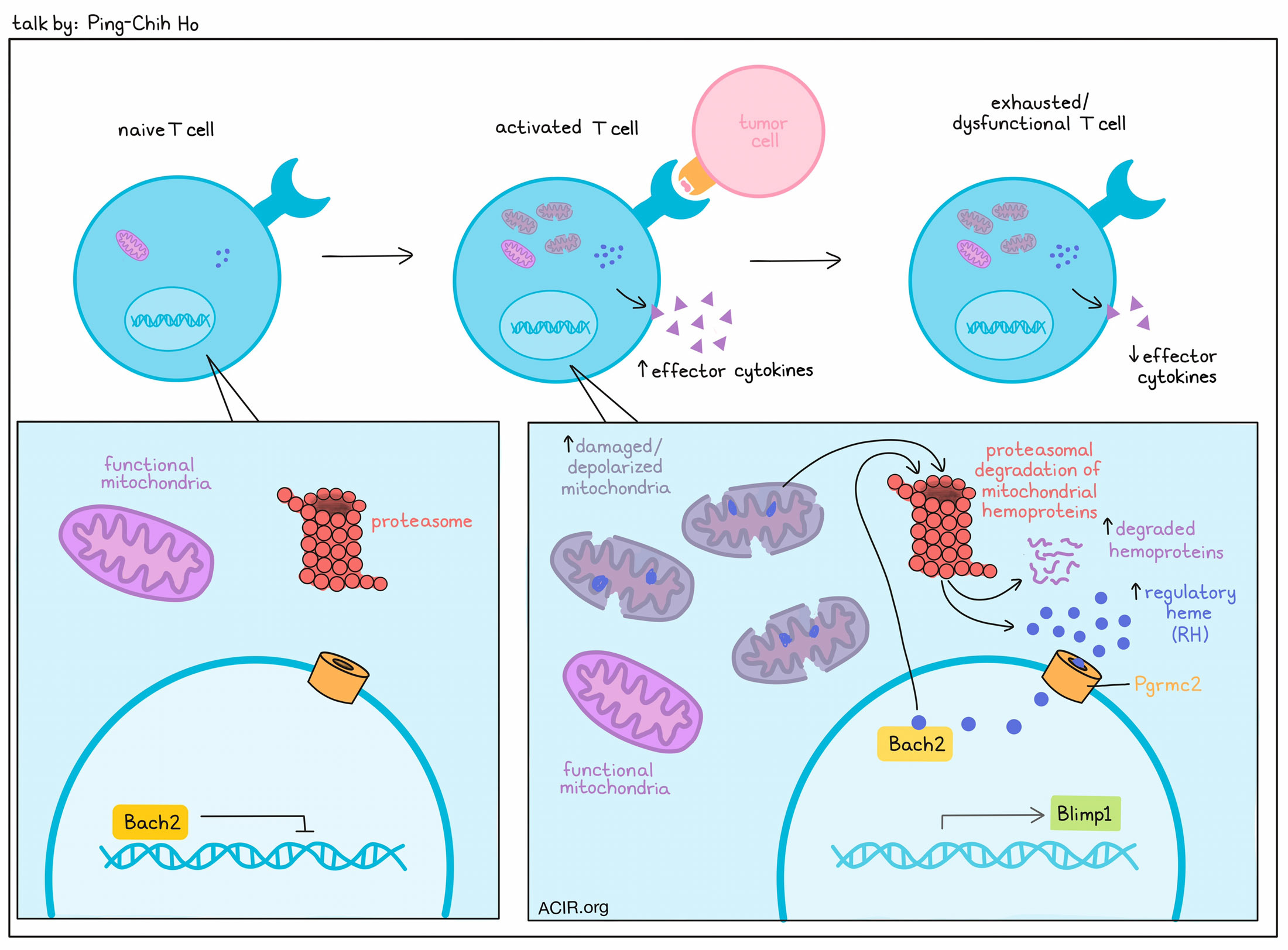
T cell exhaustion, occurring under chronic antigen exposure, is driven by epigenetic programming that becomes irreversible as it progresses. Discussing strategies to retain responsiveness in T cells, Ping-Chih Ho noted that progenitor exhausted and effector T cells show differential mitochondrial fitness. In particular, exhaustion was associated with the accumulation of depolarized/damaged mitochondria due to dampened mitophagy (the degradation of mitochondria by autophagy) despite high levels of genes related to the proteasome. Looking more closely, the researchers saw high levels of regulatory heme (RH; cytoplasmic heme released by degradation of mitochondrial hemoprotein) in exhausted CD8+ T cells with dysfunctional mitochondria. Blocking protein degradation abrogated the increase in RH, while culturing the cells in Hemin, a structural analog of heme, under conditions of repetitive stimulation induced features of terminally exhausted cells, including dampened proliferation and impaired production of effector cytokines. Transfer of these cells into mice, followed by infection with acute LCMV (Armstong) led to cells with an similar exhausted phenotype in vivo. Studying the mechanism behind this, the researchers found that heme binds to the Bach2 hemoprotein – an important repressive transcription factor for the modulation of T differentiation – leading to BACH2 degradation and reduced DNA binding. A mutant form of Bach2 that could not bind heme, on the other hand, could sustain repressive transcriptional activity. Cells expressing the mutant Bach2 did not show an infiltration advantage, but did show higher potential to sustain a progenitor phenotype and resist exhaustion. These cells also showed downregulation of Blimp1, a transcription factor needed for progression to the terminally exhausted state. Mechanistically, the researchers found that BACH2 also binds to the Blimp1 promoter to suppress Blimp1 production. Similar results were observed with knockout of Pgrmc2 (a heme chaperone that aids transport of cytoplasmic heme to the nucleus), which inhibited terminal differentiation and enhanced the polyfunctionality of TILs. Overall, these results reveal mitochondrial degradation as a key determinant of T cell exhaustion, and point to several potential targets that could limit T cell exhaustion in cancer immunotherapy.
Temporal single cell profiling identifies B-cell specific checkpoint molecules that regulate anti-tumor immunity - Lloyd Bod - Massachusetts General Hospital, Boston, Massachusetts
B cells are often overlooked in the tumor microenvironment, but Lloyd Bod has made strides towards understanding the balance between their anti- and pro-tumorigenic functions. For example, while B cells can play roles in antigen presentation, costimulation, humoral responses, inflammatory cytokine production, and the formation of tertiary lymphoid structures, they can also produce immunosuppressive cytokines, direct T cell inhibition, and promote the formation of Ig immune complexes. The relationship between infiltration of B cells in tumors and outcomes also varies across different tumors and studies. To determine whether certain B cell subsets might show more clear correlations with response or non-response, Bod isolated and characterized B cells from the tumors, draining lymph nodes, and non-draining lymph nodes of mice bearing B16F10 melanomas. This revealed a cluster of B cells that expressed TIM-1 and increased over time in the draining lymph nodes. Further characterization of these TIM-1+ B cells showed that they expressed increased levels of numerous inhibitory checkpoint molecules. Similar results were observed in samples of human melanoma and other cancers. Investigating the roles of these inhibitory receptors on B cells, the researchers performed an in vivo functional screen in which different inhibitory receptors were knocked out. While deletion of TIM-3, TIGIT, PD-1, LAG-3, or IL-10 in B cells had little to no impact on tumor growth, deletion of TIM-1 in B cells dramatically inhibited tumor growth. Mechanistic studies further suggested that TIM-1 deletion enhanced sensitivity to IFNγ, leading to enhanced activation of an antigen presentation by B cells, but not increased humoral immunity. Further, this enhanced B cell activation could promote T cells towards effector and cytotoxic, rather than regulatory phenotypes. Together, these results identify TIM-1 as an important B cell checkpoint and possible target for immunotherapies to enhance B cell-mediated support for antitumor immunity.
Identifying regulators of tumor immunity by spatial functional genomics - Brian D. Brown - Icahn School of Medicine at Mount Sinai, New York, New York
Brian Brown’s talk focused on what drives various organizational structures and conditions in heterogeneous tumor microenvironments. To better understand how cancer controls the TME, Brown and colleagues performed receptor/ligand pairing analysis, which identified 35-50 factors regulating macrophages, particularly in ovarian cancer. Given that CRISPR screens are limited to cell autonomous effects, and functional genomics approaches cannot identify cell-extrinsic phenotypes (like cell recruitment) or functions of cytokines and chemokines, the researchers developed a pro-Code/CRISPR library that would target the knockout of 35 receptors and ligands in ID8 ovarian cancer, and allow for readouts of CRISPR screens at the level of the barcoded proteins in vitro or in vivo. While none of the knockouts affected cancer cell fitness, gene knockouts in vivo recapitulated intratumoral clonal heterogeneity. Loss of Ccl7, Ccl3, and Calr were found to increase tumor burden, while loss of Paur, Mif, Serpine1, and Thbs1 were associated with reduced tumor burden. Investigating the role of CCL7 loss in supporting tumor growth, the researchers found even when CCL7 was knocked out, tumor cells outcompeted the growth of parental cells in a mixed tumor context. CCL7 knockout in tumor cells also promoted intratumoral heterogeneity, an immune-excluded TME, and clonal selection, suggesting that CCL7 is a master regulator of the immune TME. In human ovarian tumors, CCL7 correlated with better prognosis and immune infiltration. On a quest for genes that influence anti-PD-1 resistance in ovarian cancer, the researchers found that in mice with heterogeneous tumors driven by the previously described CRISPR library, loss of IL-4 made tumors more sensitive to anti-PD-1, suggesting that IL-4 promotes resistance to treatment. Further, IL-4 was found to be produced by mouse and human ovarian cancer cells, and promoted resistance to anti-PD-1 via IL-4-induced signaling in ovarian tumor macrophages that caused macrophage enrichment and T cell reduction in the TME, as well as reduced formation of TLS-like lymphoid aggregates. Importantly, the effects of both CCL7 and IL-4 were spatially restricted, leading to localized changes in the immune TME and the tumor, and driving selection and clonal outgrowth. This work thus emphasizes the importance of addressing heterogeneous tumor features and using heterogeneous tumor models.
By Lauren Hitchings, Shishir Pant, Ed Fritsch, and Ute Burkhardt



