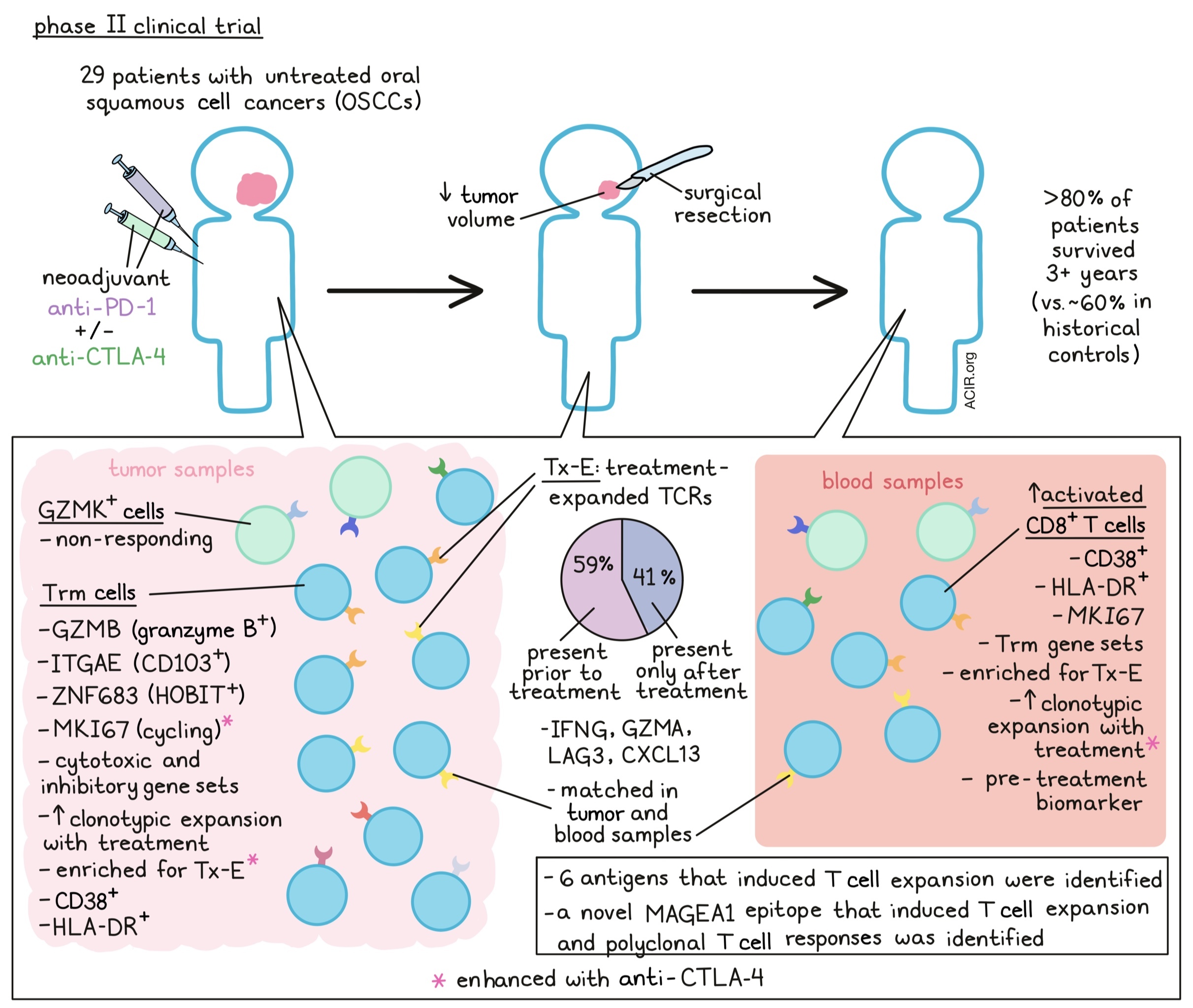
Immune checkpoint blockade (ICB) has exciting potential in treating oral squamous cell cancers (OSCCs) in the neoadjuvant setting (i.e. prior to surgery). Although the identity of the responding T cells remains to be determined, tissue-resident memory (Trm) T cells, which play key roles in long-term immunity and can be rapidly engaged for effector responses, are an intriguing possibility. Recently reported in Cell, Luoma and Suo et al. identified Trms as the major responders to neoadjuvant ICB in an OSCC clinical trial.
In this phase 2 trial, 29 previously untreated OSCC patients were given anti-PD-1 therapy alone or with anti-CTLA-4 before surgical resection (within one week of the last dose of ICB). Therapeutic outcomes were promising – the majority of patients experienced reductions in tumor volume before surgery, and >80% of patients survived out to three years, relative to ~60% in historical cohorts. To investigate T cell responses, tumor biopsies and/or blood samples were collected pre-treatment, during treatment, and after surgery for single-cell RNA and TCR analyses.
To begin, Luoma and Suo et al. investigated T cell phenotypes within the OSCC tumors using scRNAseq. CD8+ T cells clustered into two main groupings: cells expressing GZMK and cells expressing GZMB. The GZMB cluster also expressed canonical Trm genes ITGAE and ZNF683 (encoding CD103 and HOBIT, respectively), along with cytotoxic and inhibitory gene sets, indicating this as a Trm population. scTCRseq showed that although all clusters had similar clonotype sizes before treatment, this ITGAE+ cluster was enriched for greater clonotype size after treatment, indicating expansion. Although cycling (MKI67) CD8+ and CD4+ T cells were also detected, their proportions increased only in the combination treatment (anti-PD-1 plus anti-CTLA-4) group.
The team next integrated bulk TCR sequencing with the scTCRseq data to locate the intratumoral TCR clonotypes that expanded following neoadjuvant ICB, which they called “treatment-expanded” (Tx-E) TCRs. Interestingly, only <1% of total clonotypes were identified as Tx-E, which the authors attribute to the short duration between treatment and resection/analysis. Most Tx-E TCRs (~59%) were found in pre-treatment tumor samples, but the remaining ~41% were only observed in tumors after ICB, suggesting de novo priming during neoadjuvant ICB. Notably, the Tx-E clonotypes were enriched in the ITGAE/GZMB T cell cluster, both in pre- and post-treatment samples. The proportion of Tx-E cells in this cluster was also higher with anti-PD-1 plus anti-CTLA-4 than with anti-PD-1 alone. In contrast, non-responding cells largely mapped to the GZMK cluster. Altogether, these results indicate that the ITGAE-expressing cells were readily invoked by ICB for treatment response, highlighting Trms as potential early responders to ICB.
Next, Luoma and Suo et al. directly compared gene expression between Tx-E and treatment-nonresponsive CD8+ T cells in tumors. Differentially expressed genes enriched in Tx-E cells encompassed effector, tissue resident, cytotoxic, and inhibitory pathways, and largely mapped to known Trm gene sets. A similar gene signature differentiated responding and non-responding CD4+ T cells; both CD8+ and CD4+ Tx-E T cells expressed effector and inhibitory genes including IFNG, LAG3, GZMA, and CXCL13. The researchers then developed a Tx-E gene signature, which they found correlated positively with treatment response and OS in an independent cohort of patients with urothelial cancer who were treated with anti-PD-L1.
To pinpoint the specific antigen targets of the Tx-E TCRs, the authors used TSCAN to screen 80 patient-derived CD8+ T cell TCRs against target cells presenting diverse self, mutant, and viral protein fragments on MHC-I. Through a GZMB reporter assay, six antigens were found targeted by TCRs that expanded after ICB treatment. These targets were a mix of self and tumor-associated antigens. Intriguingly, the team identified a novel epitope of the cancer-testis antigen MAGEA1 that induced a polyclonal response, which could have potential as an immunotherapy target.
Having analyzed intratumoral responses to neoadjuvant ICB, the researchers next considered whether these TCRs mapped to systemic T cell responses. In the blood, a CD8+ T cell cluster expressing CD38, HLA-DRA, and MKI67 increased in frequency over time. Interestingly, tumor Tx-E TCRs mapped largely to this population, and the previously identified tumor ITGAE/GZMB cluster also highly expressed CD38 and HLA-DR. Furthermore, TCR sequences from CD38+ HLA-DR+ CD8+ T cells sorted from blood matched tumor TCRs. Overall, the concordance between tumor and blood T cell phenotypes and TCR clonotypes responding to therapy suggests both a local and systemic response to ICB.
Because a significant portion (~41%) of Tx-E TCRs in the tumor was only detected post-therapy, the team investigated dynamics of these clones in the blood. The majority of these emergent TCRs were also found in blood after ICB, expanding ~2 weeks into treatment and declining by 10-12 weeks. These cells, and in fact all Tx-E clonotypes, expanded most significantly in the blood with the combination of anti-PD-1 and anti-CTLA-4 compared to anti-PD-1 alone – an interesting result given the known role of anti-CTLA-4 in supporting T cell priming.
Given the concordance between blood and tumor TCR clonotypes and phenotypes, the team finally investigated whether blood T cell features could potentially serve as biomarkers for ICB response. Strikingly, the CD38+ HLA-DR+ CD8+ T cell population in pre-treatment blood samples correlated with treatment response. These cells expanded during the course of treatment and displayed proliferative (Ki-67) and inhibitory (PD-1) markers, suggesting this activated T cell population as a potential biomarker for ICB efficacy. Pre- and on-treatment PD-1+ KLRG1- cells also strongly correlated with pathological response.
In conclusion, Luoma and Suo et al. identified Trm CD8+ T cells as early responders to neoadjuvant ICB in OSCC patients. Further investigating T cell phenotypes and responding populations could better tailor immunotherapy selection and delivery in future iterations. The identification of CD38/HLA-DR as potential T cell biomarkers can support this approach. Finally, the identification of a novel epitope capable of a CD8+ T cell response in OSCC provides another target for immunotherapy applications.
Write-up by Alex Najibi, image by Lauren Hitchings
Meet the researcher
This week, lead author Kay Wucherpfennig answered our questions.

What was the most surprising finding of this study for you?
We investigated which immune cells respond early to pre-surgical (neoadjuvant) immune checkpoint blockade. The surprising finding was that those T cells that responded by clonal expansion expressed a tissue-resident memory program. We also found that there is an early systemic response to neoadjuvant immune checkpoint blockade characterized by a population of highly activated, proliferating T cells. Thus, neoadjuvant checkpoint blockade induces a local response in the tumor as well as a systemic response. We think that the systemic response may be important in protecting against the outgrowth of metastases.
What is the outlook?
We identified a novel biomarker in these studies. A substantial fraction of our patients had a pathological response (tumor necrosis evaluated by histology). We found that the activation state of circulating PD-1+ T cells was an important biomarker of pathological response. Importantly, the percentage of PD-1+ T cells was not associated with response, but the fraction of PD-1+ cells that were negative for the terminal differentiation marker KLRG1 strongly correlated with pathological response. We are now investigating the hypothesis that tissue-resident memory T cells are critical for cancer immunotherapy. This is relevant for many approaches to immunotherapy, including CAR T cells.
What was the coolest thing you’ve learned (about) recently outside of work?
I am riding the Pan-Mass Challenge for Dana-Farber beginning of August. It’s 192 miles from Sturbridge to Provincetown at the tip of Cape Cod.



