While the effect of PD-1 signaling in T cells in response to binding with PD-L1 has been reasonably well established, PD-L1 signaling via its cytoplasmic domain, so-called PD-L1 reverse signaling, has not been broadly studied. In tumor cells, PD-L1 reverse signaling protects the cells from IFN-induced cell death, but its effects on antigen-presenting dendritic cells (DCs), the key cell type involved in immune priming, are unknown. Lucas et al. explored this mechanism using in vivo and in vitro models in their current publication in Cell Reports.
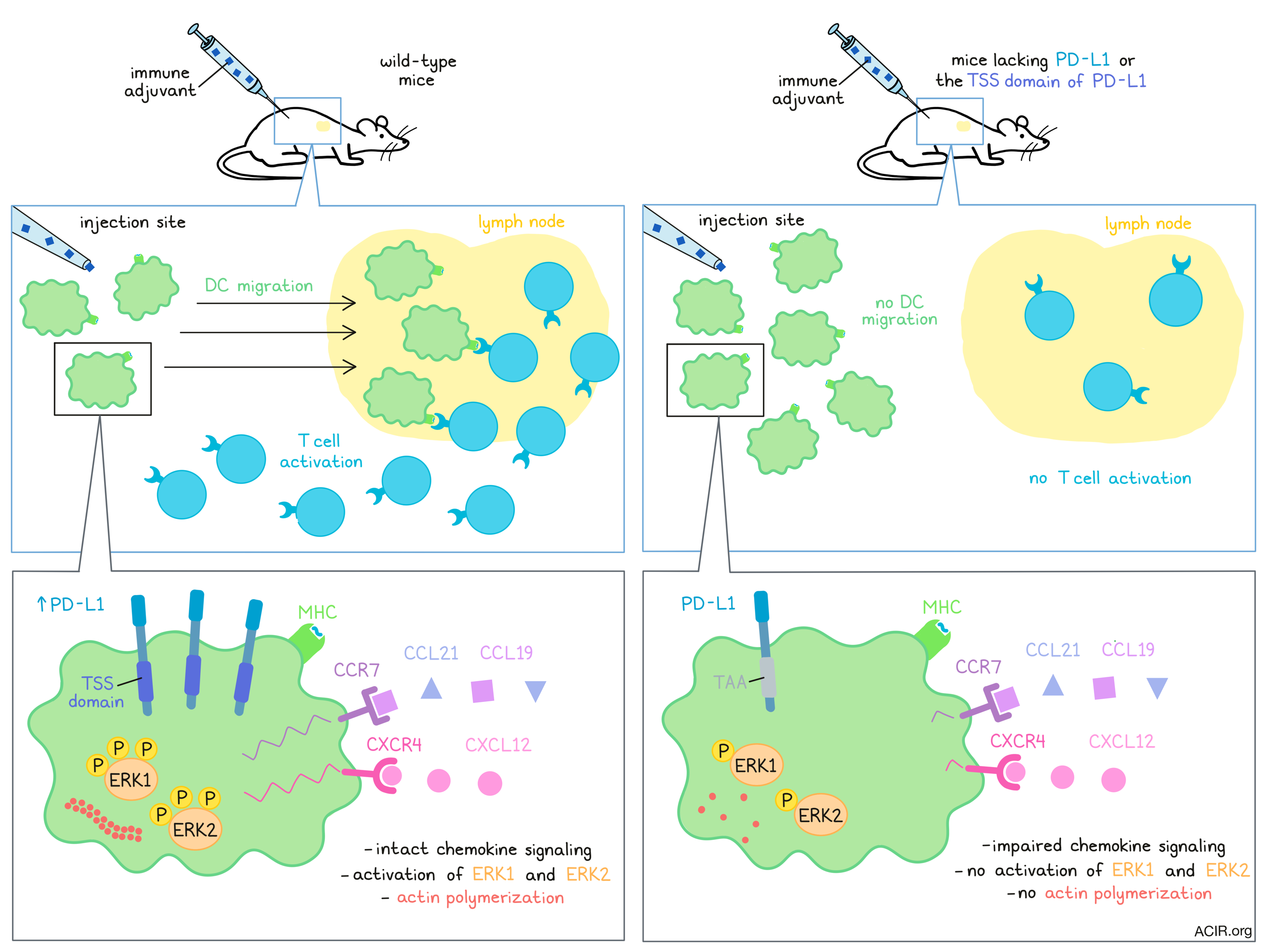
Lucas et al. started by researching immune cell compositions in wild-type (WT) and PD-L1-/- mice, at baseline and following intradermal or subcutaneous injection with the immune adjuvant poly(I:C) to induce IFN signaling. Untreated WT and mutant mice both had similar cell frequencies and cell types in the draining lymph nodes (dLN), and after injection of WT mice, both conventional dendritic cell subsets, cDC1 and cDC2, upregulated PD-L1. However, the researchers found significantly fewer cDC1 and cDC2 in the dLN of the treated mutant mice. The opposite effect was seen in the skin at the site of injection, where more DCs were present in the PD-L1-/- mice.
To evaluate whether these differences were due to limited DC migration in the knockout mice, the researchers irradiated WT mice and reconstituted them with WT or PD-L1-/- bone marrow. Using a FITC painting assay to label migratory DCs in the skin, the migration of the DCs from the skin site of FITC injection to the dLN could be followed. Both mouse types showed migrating DCs 24 hours after FITC painting. However, after poly(I:C) injection, there were significantly more migrating DCs in the WT mice than in the mice reconstituted with PD-L1-/- bone marrow. The authors then co-transferred (1:1) differentially marked and labeled bone marrow-derived DCs (BMDCs) from WT and PD-L1-/- mice into the footpad of WT mice that had been injected with poly(I:C). Analysis 24 hours later showed a significant decrease in the ratio of PD-L1-/-:WT BMDCs in the dLN and an increase in the ratio of PD-L1-/-:WT BMDCs in the skin. No differences in viability were found, indicating that the effects were due to migration differences.
Based on previous research, the authors examined whether the threonine-serine-serine (TSS) region of the cytoplasmic PD-L1 tail controlled DC migration, by mutating the three amino acids to alanine-alanine-alanine using CRISPR-Cas9 in C57BL/6 mice embryos. Using the resulting inbred mice (Pdl1CyMt ), the researchers could assess the effects of the intracellular region of PD-L1, given that in these mice, the extracellular domain of PD-L1 remains intact, in contrast to the PD-L1-/- mice. The previous experiments were repeated, and again the mutant mice had less migration of DCs from the skin to the dLN. The authors did not find any upregulation of PD-L1, nor any differences in CD80, CD86, CD40, or MHC-II expression between WT and Pdl1CyMt mice, suggesting that the limitations in DC migration were not dependent on CD80 or CD86 expression. These results were confirmed by co-transfer of WT and Pdl1CyMt BMDCs into WT mice after injection with poly(I:C). These findings suggest that PD-L1 intracellular signaling required the TSS region for DC migration.
To assess whether T cell responses were affected, the researchers immunized mice subcutaneously with ovalbumin (ova)/poly(I:C)/aCD40, which did not require DC migration, as the soluble antigen could transit to the LNs via the lymphatic vessels. No differences were found in activated CD8+ T cells between both mutant and WT mice. Next, the authors infected mice intradermally with Listeria monocytogenes-expressing ovalbumin (LM-ova); since bacteria are too large to transit through the lymphatic capillaries, a T cell response would require the help of migratory DCs. Again, antigen-specific CD8+ T cells were activated in the dLN of WT mice, but not, for this antigen, in the mutant mice. To eliminate the possibility that this was due to a PD-L1 reverse signaling defect in T cells, WT OT-I T cells were transferred into mutant mice, resulting in a similar loss of T cell priming. No loss of priming was observed when LM-ova was injected systemically, and in vitro experiments showed that WT and mutant (PD-L1-/- and Pdl1CyMt ) BMDCs were capable of processing and presenting antigens to T cells. These data together suggest that the differences found in mutant mice were due to inhibition of DC migration to the dLN.
To dive deeper into the causative factors of defective migration, the researchers used transwell experiments to assess chemokine signaling. While activated WT BMDCs migrated to CCL21 and CCL19, both signaling through CCR7, the BMDCs from the mutant mice did not. A similar effect was found for CXCL12, which signals through CXCR4. S1P-mediated migration, on the other hand, was not interrupted in the mutant mice. Confirming that the chemokine effects were not caused by missing receptors, the researchers found that CCR7 was upregulated upon activation on BMDCs and the cDCs from the mice of all genotypes. This suggests that signaling downstream of the receptor was responsible for the lack of migratory activity in DCs deficient in intracellular PD-L1 signaling. Chemokine receptors are G-protein coupled receptors and consistent with this, Lucas et al. found that WT BMDCs increased phosphorylation of ERK1 and ERK2 upon activation while mutant BMDCs did not. Actin polymerization was also inhibited in the Pdl1CyMt BMDCs. AKT phosphorylation was not affected, indicating that reverse PD-L1 signaling specifically impacted the Gα arm of the G-protein pathway. These effects were confirmed in in vivo skin DCs.
In conclusion, Lucas et al. showed that intracellular PD-L1 signaling was essential for activated DCs to migrate to the LN. A lack of PD-L1 reverse signaling affected chemokine receptor signaling, directly impairing the migration of DCs. It is currently unknown whether ligand binding or antibody blockade of the PD-1 axis affects this signaling pathway in DCs. These questions are vital to study to elucidate the impact of PD-L1 reverse signaling on DC migration and T cell activation.
Write-up by Maartje Wouters, image by Lauren Hitchings.
Meet the researcher
This week, first author Erin Lucas answered our questions.

What prompted you to tackle this research question?
We first grew interested in PD-L1 expression by lymphatic endothelial cells and found that inflammatory signals increased PD-L1 expression during infection. We noted that PD-L1 not only responded to the inflammatory environment, but impacted lymphatic endothelial cell function. This was extremely intriguing to us, especially given the importance of PD-L1 immunotherapy in the clinic on treating patients with cancer. Following our work examining the role of PD-L1 in lymphatic endothelial cells, we were very interested in exploring why type 1 IFN produced during infection increased PD-L1 expression at such early time points. We began by characterizing the immune cell populations in the lymph node one day after subcutaneous or intradermal polyI:C injection and found a marked decrease in the number of DCs in the LN in the PD-L1 knockout mouse. As we know how important DC antigen presentation is for the initiation of immune responses, we felt understanding how loss of PD-L1 could affect DC migration was important to understand, especially given that PD-L1 blocking antibodies are currently being used in the clinic. We believed that fully understanding PD-L1 reverse signaling could have implications for patients undergoing PD-L1 immunotherapy.
What was the most surprising finding of this study for you?
The most surprising finding of our study for me is the profound impairment in DC migration following an immune stimulus in the Pdl1CyMt mouse model. Not only was it striking just how much of an impact making a three amino acid mutation in the short cytoplasmic tail of PD-L1 could have on DC migration, I am very intrigued as to why we only observe this difference in an inflamed environment, and hope to investigate this further.
What was the coolest thing you’ve learned (about) recently outside of work?
I have always been interested in Mediterranean history and antiquity. I was fascinated to learn that both ancient Greeks and ancient Egyptians discovered and documented fossils, and recorded their findings. In Ancient Egypt, these fossils were collected and revered as sacred relics of the god Set, and were placed in tombs and shrines. In Greece, it is thought that many mythical beasts, including griffons and cyclops, were inspired by the discoveries of large fossilized bones. Learning this has reminded me that across all time and civilization, humanity has always tried to study and explore our world, and to explain the world as we understand it. As a scientist, it is extremely gratifying to know that I can not only form hypotheses to explain the data I get, but I am then able to test my hypotheses and contribute to the wealth of knowledge in our world



