In the setting of colorectal cancer (CRC), mismatch repair (MMR)-deficient or microsatellite instability (MSI)-high disease tends to have higher tumor mutation burdens (TMB) and better responses to immunotherapy than MMR-proficient and microsatellite stable (MSS) disease. However, MSS CRC still has a mutation burden that is comparable to other cancers that respond to immunotherapy, suggesting that other factors may contribute to lack of response. To better study CRC, Westcott et al. developed a mouse model that mimicked the microenvironment of human disease and enabled the tracking and characterization of defined antigen-specific T cells. Using this model, the team evaluated expression levels of neoantigens in low-TMB CRC; the results were recently published in Nature Cancer.
To develop a better mouse model for CRC, Westcott et al. first developed a neoantigen prediction pipeline integrating HLA haplotype calling and affinity prediction pipeline algorithms and applied it to TCGA and other datasets. This showed that compared to patients with MSI-high CRC, patients with MSS CRC had lower mutation burdens, lower average predicted neoantigen clonality, and lower average expression of genes encoding clonal predicted neoantigens. Still, organoids derived from patients with MSS CRC presented bona fide neoepitopes, and potentially others that were below the threshold for detection, suggesting that these patients may harbor actionable neoantigens at low levels.
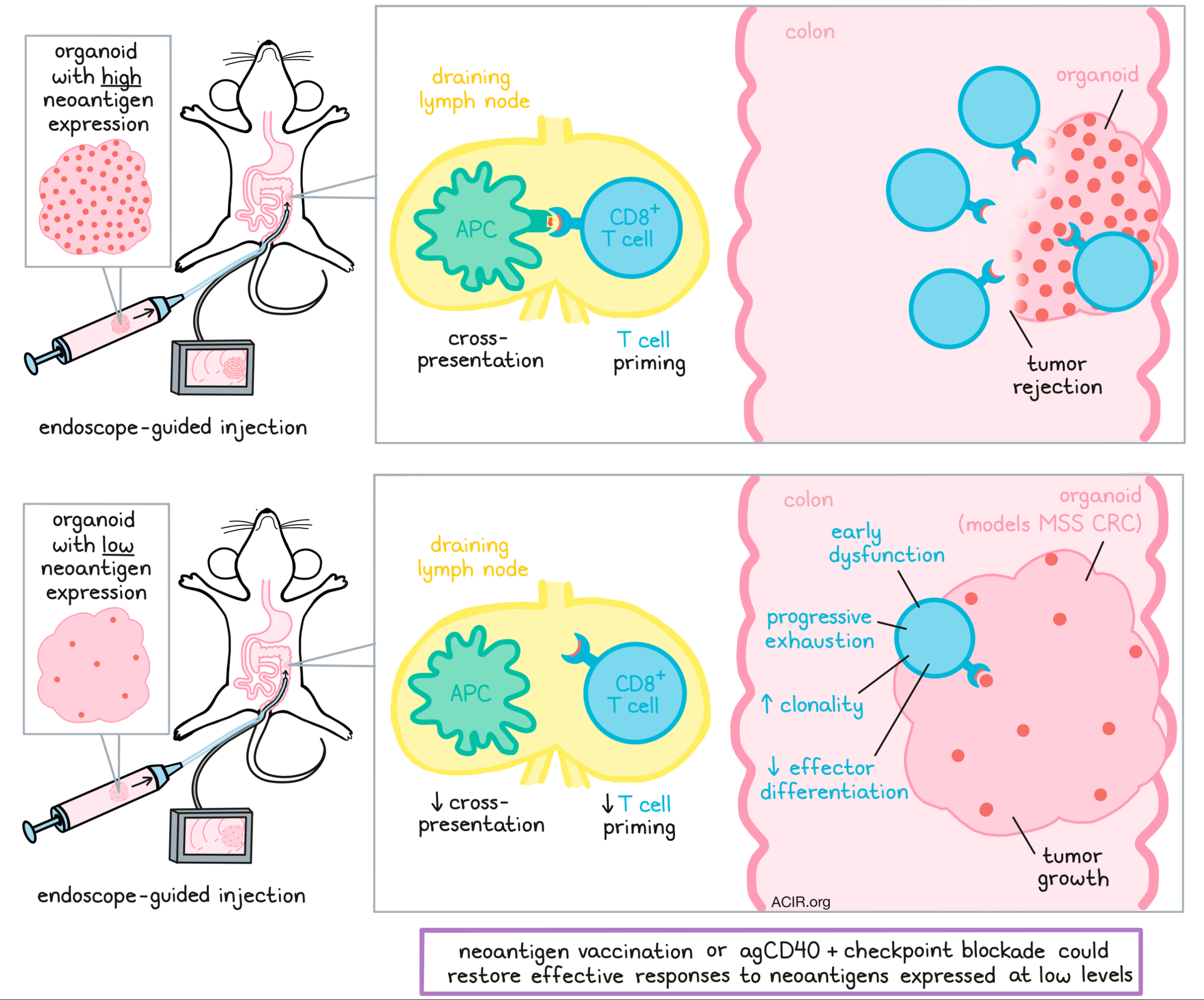
To test whether neoantigens could be targeted at low levels, Westcott et al. developed organoids that modeled common genetic mutations in MSS CRC, and maintained distinct levels of antigen expression throughout tumor development. Within this flexible system, the team studied organoids with a 400-fold range in expression of the OVA epitope SIINFEKL, which correlated directly with presentation of the epitope on MHC-I and served as a model neoantigen.
When organoids were transplanted into the colons of mice (guided by endoscopy), control organoids that did not express the model neoantigen grew into large tumors, while those with high expression of the model neoantigen were fully rejected. Organoids that expressed intermediate levels of the neoantigen formed small tumors, while those that expressed the lowest levels of neoantigen grew to about the same size as controls. The tumors with low neoantigen expression had a “cold” immune environment with minimal T cell infiltration. Interestingly, these tumors did show infiltration of neoantigen-experienced and SIINFEKL-specific T cells, but did not undergo immunoediting and maintained a low level of neoantigen expression.
Investigating how tumors with low expression of a high-affinity neoantigen manage to escape immune rejection, the researchers compared T cell responses in low-antigen versus high-antigen tumors and found that in low-antigen tumors, T cell responses were delayed and were lower in magnitude, and a higher portion of antigen-specific T cells from antigen-low tumors and LNs were TCF1+GZMB- rather than TCF1-GZMB+, suggestive of impaired effector differentiation. There was also a higher portion of TCF1-GZMB- T cells, indicative of dysfunctional cells lacking both progenitor and effector functions. These effects became more pronounced with time, and by day 14, more cells also showed expression of PD-1, TIM3, LAG3, and 2B4. In a separate long-term analysis that extended through day 42, the portion of terminally exhausted TCF1-TIM3+ increased, the population of progenitor cells decreased, and fewer T cells were polyfunctional.
Looking more closely at the functionality of T cells responding to low-antigen tumors, the researchers used an in vivo killing assay with SIINFEKL-loaded cells. This showed that T cells from high-antigen tumors were cytotoxic against target cells, while T cells from low-antigen tumors were less cytotoxic, with each antigen-specific T cell killing fewer target cells. T cells from low-antigen tumors were also more clonal. Together these results suggested that low expression of neoantigens induced T cell responses that were immediately dysfunctional and became progressively exhausted.
Given that impaired effector differentiation and early dysfunction are often products of poor T cell priming, Westcott et al. explored this hypothesis. After showing that impaired priming was not the result of a lack of T cell help, they found that cDC1s and other antigen-presenting cells exposed to low-antigen tumors were less capable of promoting proliferation and effector differentiation through cross-presentation. Since T cells primed in vitro were capable of killing low-antigen organoids, the researchers concluded that low neoantigen expression limits T cell cross-priming, but not T cell recognition of antigen on tumors. In vivo findings supported this, as injection of high-antigen organoids followed by low-antigen organoids, co-injection of high-antigen and low-antigen organoids, or transfer of in vitro-primed T cells with low-antigen organoids all rescued T cell priming and induced strong responses against low-antigen tumors.
In an effort to investigate therapeutic strategies to potentially rescue T cell responses in MSS CRC, the researchers investigated the use of a neoantigen vaccine to enhance priming in mice with established tumors expressing low levels of neoantigens. In this setting, a prime and boost of an OVA peptide-based vaccine increased antigen-specific T cells in peripheral blood and induced tumor regression, including several complete responses in mice.
Investigating more readily available antibody-based therapies, the researchers also tested agonist CD40 (agCD40), anti-PD-1, anti-CTLA, and various combinations of the three. As monotherapies, each treatment induced some complete responses in antigen-low tumors, though the responses induced by agCD40 were more durable and controlled metastasis better than either checkpoint blockade therapy. Compared to the monotherapies, all combination therapies were superior, again with agCD40-containing therapies imparting the most control. Finally, the researchers tested an adoptive cell transfer regimen and found that ex vivo-activated cells delayed tumor growth and controlled metastases, but only induced one complete response. Importantly, organoids that escaped immunotherapy were still sensitive to antigen-specific T cell-mediated killing, suggesting that tumors that have escaped prior immunotherapy could still be treated if T cell priming against poorly expressed neoantigens could be rescued.
Overall, Westcott et al. showed that patients with MSS CRC with a low mutation burden may still harbor neoantigens at low levels that can lead to tumor control. Using their novel mouse model, they found that when neoantigens are physiologically expressed at low levels, cross-priming of T cells is impaired, leading to immediate dysfunction and progressive exhaustion. Restoring T cell priming through immunotherapy, particularly agCD40 and vaccine therapy, could potentially pharmacologically generate effective responses to neoantigens and thereby improve patient outcomes.
Write-up and image by Lauren Hitchings
Meet the researcher
This week, first author Peter Westcott answered our questions.

What prompted you to tackle this research question?
We were intrigued by the remarkable efficacy of immune checkpoint blockade in patients with DNA mismatch repair-deficient colorectal cancer, but wanted to better understand why most colorectal cancer (mismatch repair-proficient) is refractory to these treatments. We wondered if there are any features of mismatch repair-proficient colorectal cancer that we could exploit to improve immunotherapy response, or if these tumors simply lack neoantigens and meaningful T cell responses altogether. Beyond the human sequencing analyses we performed, we wanted to be able to study neoantigen-specific T cell responses at fine resolution in a model that really captures the microenvironment, genetics, and histopathology of colorectal cancer in humans. We could not find a model that fit this bill, so we set out to make one.
What was the most surprising finding of this study for you?
What we found most surprising was the wildly different effects of neoantigen expression level on the tumor-specific T cell response depending on the particular stage of T cell development. At initial T cell activation, neoantigen expression is limiting for cross-priming, and really tunes the quality of the resulting T cell response. However, at the effector T cell stage, following therapies that boost priming, neoantigen expression was no longer limiting, and low expression mediated potent tumor cell killing. This has important therapeutic implications, as immune “cold” tumors may harbor neoantigens that never primed productive T cell responses and thus fly under the radar in the sense of detection by functional assays for tumor-infiltrating lymphocytes. But if priming can be rescued, these same neoantigens may be sufficient to render tumors vulnerable to killing.
What was the coolest thing you’ve learned (about) recently outside of work?
I definitely do not have a green thumb and have been struggling to keep this grocery store phalaenopsis orchid alive for 2 years. I have battled with fungus gnats, root rot, too much light, too little light, and fungus gnats... always the fungus gnats! Finally, after a new pot and fancy medium, it seemed to be thriving. All was great, until I noticed a filamentous white mold emanating from the succulent green roots. How could this be?! But my chagrin gave way to curiosity after some scientific consultation with Google. These were actually root hairs, which orchids use to secure themselves to the bark of trees in the wild. Much cooler than mold!




