
Last week, the ACIR team attended the AACR Tumor Immunology and Immunotherapy conference that virtually took place in Boston, MA. This week’s extensive special feature covers select sessions from the conference. We have organized the content by topics below.
- Approaches to T cell priming and vaccines
- Regulators of tumor immunity
- Myeloid Compartment in the TME
- CAR cell therapy
- NK cells
Approaches to T cell priming and vaccines
A case for priming, not checkpoint - Robert H. Vonderheide, Abramson Cancer Center of the University of Pennsylvania, Philadelphia, Pennsylvania
In one of two keynote presentations on the opening day of the conference, Robert H. Vonderheide from the Abramson Cancer Center of the University of Pennsylvania spoke with optimism regarding recent drops in cancer death rates, which are at least partly attributed to advancing immunotherapies. However, since the advent of CTLA-4 and PD-1/PD-L1 blockades, no new checkpoint blockades have been made available for use. In an effort to break through this apparent ceiling, Vonderheide turned to recent data showing that while high tumor mutation burdens and identified neoepitopes predict response to checkpoint blockade, they do not correlate strongly with an inflamed T cell signature (though this also independently correlates with response to checkpoint blockade). To explore this further, Vonderheide and colleagues used a genetically engineered KPC mouse model of spontaneous pancreatic ductal adenocarcinoma (with mutant Kras and mutant p53) that is resistant to single-agent anti-PD-1 or anti-CTLA-4. In this model, they identified early, systemic, and progressive dendritic cell dysfunction, and a low frequency of cDC1s in tumors or regional lymph nodes. The functional deficit was found to be linked to high levels of IL-6 and IL-6-induced apoptosis. When IL-6 was neutralized with antibodies, cDC1 frequency and function were restored. cDC1 function could also be restored with CD40 agonism, with further benefit from the addition of FLT3 ligand, which increased the numbers of cDC1s and their maturation. Given the ability of CD40 agonism to enhance T cell priming, Vonderheide and colleagues investigated whether CD40-agonizing mAb pretreatment prior to chemotherapy or radiation might act as a vaccine. Several years ago this strategy was tested in chemotherapy-naive patients with metastatic pancreatic carcinoma, and the addition of CD40 agonistic antibodies to standard-of-care gemcitabine chemotherapy appeared to induce more of a response than what would be expected with chemotherapy alone. Interestingly, when this strategy was explored in mice, the effect was found to be independent of T cells, and instead depended on CD40 activation on macrophages. However, the addition of abraxane chemotherapy to agonist anti-CD40 and gemcitabine did induce an antitumor T cell response, as well as a shift in the myeloid compartment from M2-like to M1-like phenotypes. With a role for T cells in this setting, Vonderheide and colleagues initiated a clinical trial, the PRINCE trial, in patients with metastatic pancreatic ductal adenocarcinoma. All patients received standard-of-care gemcitabine and abraxane (nab-paclitaxel), and one of two doses of agonist anti-CD40. Half the groups received additional treatment with nivolumab. The majority of patients achieved durable, long-term remissions, and the overall response rate was increased compared to what would be expected from chemotherapy alone. A phase II clinical trial is ongoing. Based on these results, Vonderheide makes the case for turning more attention towards priming, rather than checkpoint blockade, to improve antitumor responses.
Engineering artificial antigen presenting cells, aAPC, for cancer immunotherapy: From bench to bedside - Jonathan P. Schneck, Johns Hopkins University School of Medicine
Aiming to improve access to and reduce the cost of autologous, antigen-specific endogenous T cell therapies from patient samples, Jonathan P. Schneck of Johns Hopkins University School of Medicine described the development of artificial antigen-presenting cells (aAPCs). He and his team used a reductionist approach by including only the minimal components required: a peptide:HLA complex and a CD28-stimulating antibody coupled to a paramagnetic nanoparticle to provide surface-mocking valency. The paramagnetic beads allow ‘enrichment’ of target-specific T cells, in addition to the ‘expansion’ effect as aAPCs. Stimulation of both healthy volunteer- and patient-derived T cells with MART-1-loaded aAPCs showed comparable expansion and phenotypes of the resulting T cells, including a central and stem-like cell memory phenotype and polyfunctionality. Multiple antigens could be used simultaneously, as evidenced by work done by NexImmune, who prepared an AML-targeting product with five peptides representing three different tumor-associated antigens (PRAME, WT1, and cyclin) and found that 38% of the stimulated T cells were antigen-specific. To increase flexibility and ease of preparation, Schneck demonstrated that aAPCs could be prepared unloaded and subsequently loaded with peptide, even in a microplate format. Using these unloaded aAPCs, he was able to show that loading peptides eluted with mild acid from surface HLA of B16 melanoma cells expressing the SIY antigen would allow stimulation of SIY-specific T cells as efficiently as aAPCs loaded with pure SIY peptide. This occurred despite the observation that the SIY peptide represented only one-millionth of the mass of loaded peptide – an intriguing observation reflecting the sensitivity and specificity of the T cell. Additional modifications to the platform (nanobead size and additional Th1-skewing cytokines) have allowed the use of aAPC to enrich and expand CD4+ T cells with both cytolytic and CD8+ T cell helper functions.
Building improved personal cancer vaccines - Catherine J. Wu, Dana Farber Cancer Institute, Boston, Massachusetts
With a focus on neoantigens, Catherine Wu of the Dana Farber Cancer Institute discussed how therapeutic cancer vaccines can be seen as an important adjunct to other therapies. Neoantigen-targeted vaccines can serve to steer an immune response against tumor cells by generating and expanding a tumor-specific T cell response with reduced toxicity towards normal, healthy tissue. In recent years, advances in next-gen sequencing approaches and HLA-binding prediction algorithms have allowed for the development of personalized vaccines specific for neoantigens that are only present in an individual patient’s cancer. Clinical studies in the adjuvant setting of high-risk melanoma and newly diagnosed glioblastoma have shown therapy with neoantigen-targeted vaccines (NeoVax) to be feasible, safe, and able to generate specific T cell responses; they also show signs of synergy with checkpoint blockade. Looking back at cryopreserved samples from patients who achieved durable responses to NeoVax with or without anti-PD-1 that persist to this day, Wu and her team identified transcriptional clusters and noted patterns in which T cells advance from naive, to cytotoxic, to memory states with little evidence of exhaustion. Further, as this pattern played out, dominant clonotypes persisted and T cell repertoires broadened, suggestive of epitope spreading. Current samples from the same patients show neoantigen-specific T cells persist long-term. In the case of a patient who had a recurrence shortly after vaccination, and a subsequent complete response on anti-PD-1 therapy, clonotypes that arose during vaccination overlapped with T cell clonotypes found in the relapsed tumor and were persistent for nearly a year. While personalized vaccines have led to impressive long-term responses in some patients, and several clinical trials are currently underway, there is still work to be done to improve neoantigen vaccines. In addition to enhancing neoantigen prediction, optimizing timing of combination treatments, and lowering costs, Wu and her team are looking for ways to target new classes of neoantigens, including unannotated ORFs, splice variants, and gene fusions. They are also considering ways to increase HLA expression within tumors, in order to reveal hidden neoepitopes. Furthermore, in an ongoing clinical trial of renal cell carcinoma, NeoVax is being locally administered with subcutaneous anti-CTLA-4. The induction of robust circulating neoantigen-specific T cell responses shows early promise, and tissue samples are being taken to understand vaccine uptake by APCs in the skin. With numerous collaborative efforts underway in neoantigen vaccine research across Boston and around the globe, Wu expects to see significant progress in this field in the coming years.
Regulators of tumor immunity
Informing therapy by elucidating natural mechanisms of tissue immunosurveillance - Adrian C. Hayday, The Francis Crick Institute, London, England
Beyond PD-1 and CTLA-4, T cells engage tissues through a variety of molecular pathways. Adrian Hayday from the Francis Crick Institute illustrated how mechanisms of tissue-specific immune surveillance may support cancer immunotherapies. γδ T cells have multiple properties that favor adoptive T cell therapy applications, including high cytotoxicity and the absence of MHC restriction, which allow allogeneic use without safety concerns due to graft-vs-host disease. γδ T cells are highly conserved between species, reside primarily within peripheral tissues, and have been shown to curtail epithelial malignancies. In the skin, γδ T cells clustered with epithelial cells expressing the protein Skint-1, a B7 superfamily member with a similar structure to PD-L1 and tissue-specific butyrophilin-like proteins (BTNLs; a class of molecules involved in γδ T cell regulation). Skint-1 was additionally expressed in the thymus, where it engaged progenitor γδ T cells to induce a selection signature noted by molecules such as CD69 and 4-1BB, along with constitutive γδ TCR activation. These selected γδ T cells could then migrate to the skin, interact with Skint-1-expressing epithelial cells, and self-renew. Because Skint-1 is known to be downregulated by trauma, the researchers questioned how γδ T cells would respond to UV irradiation without prior Skint-1 signaling. Antibody blockade of Skint-1 diminished the γδ T cell selection signature and responsiveness to UV irradiation, increasing skin damage and carcinogenesis. Additionally, these γδ T cells could be boosted through a 4-1BB agonist, increasing TCR activation and effector molecule expression – an effect blocked by anti-Skint-1 antibody treatment. Thus, Skint-1 in the steady state maintains a differentiated state of γδ T cells with the capacity to respond to costimulation and carcinogen-induced damage. In a final insight, Hayday characterized the γδ TCR-BTNL interaction as proceeding in both a non-clonotypic (i.e. conserved and innate) manner in response to these conserved epithelial regulators, as well as in a classical adaptive response through antigen stimulation of the clonotypic TCR: an innate-like monitoring in the steady-state and adaptive response during stress and carcinogenesis – ‘adaptate’.
Discovery of new immunotherapy targets and mechanisms leveraging CRISPR - Arlene H. Sharpe, Harvard Medical School, Boston, Massachusetts
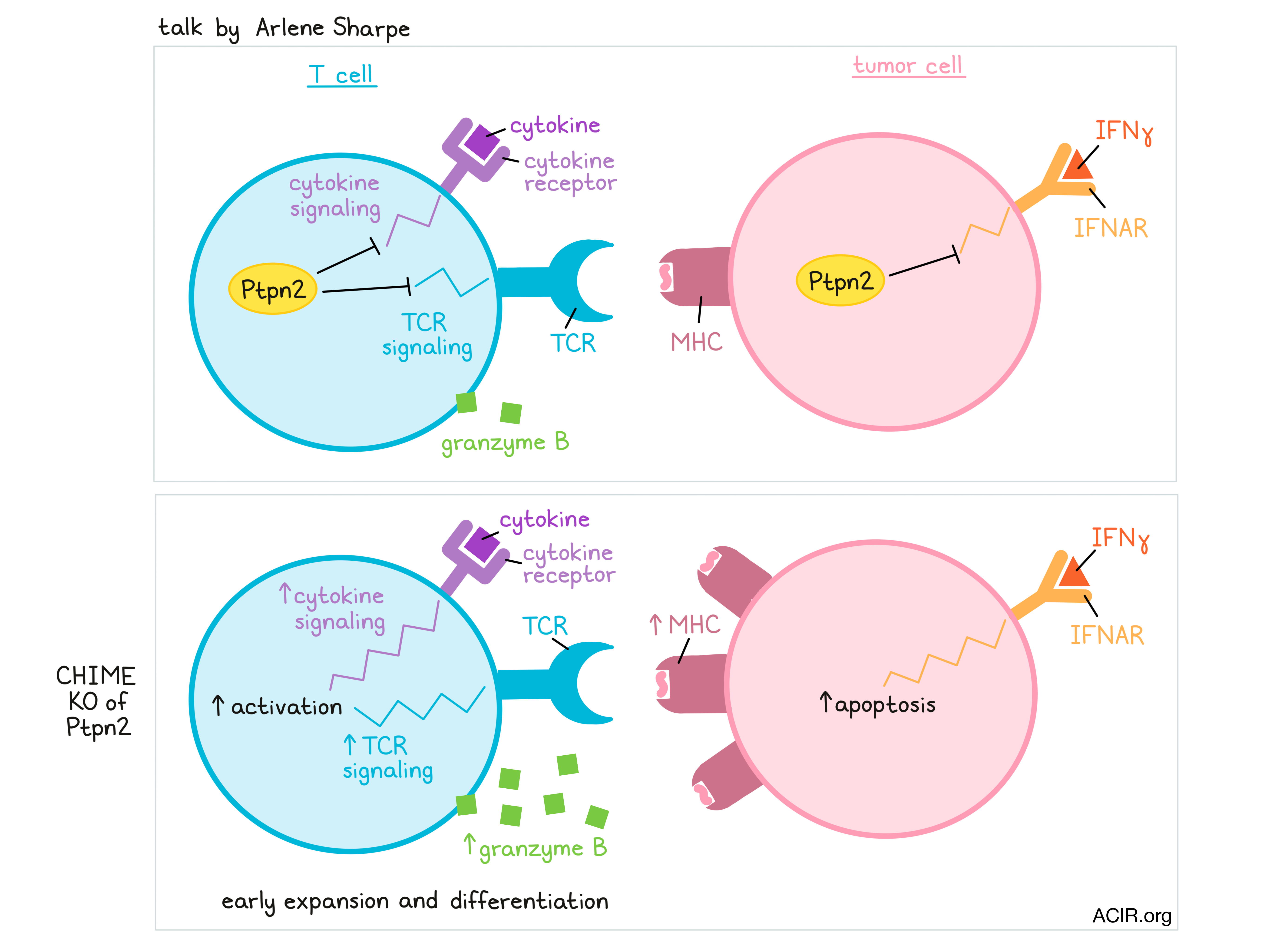
In her talk, Arlene Sharpe of Harvard Medical School discussed the development of CHIME – a CRISPR-based platform that allows researchers to knock out genes in immune lineages (B cells, naive lymphocytes, CD4+ T cells, CD8+ T cells, macrophages, and DCs) in vivo, without affecting immune homeostasis or function. CHIME can even be used to evaluate genes in a pooled fashion and is being developed to knock out two genes simultaneously in the same cell. This system allows for the identification of immune cell-intrinsic effects and known and novel regulators of antitumor responses. In one screen, a novel regulator, Ptpn2, caught Sharpe and her colleagues’ attention. Ptpn2 is a phosphatase that plays a role as a negative regulator of TCR signaling and cytokine signaling in T cells, and is associated with autoimmunity in both mice and humans. Further, it also plays a cell-intrinsic role in tumor cells, regulating IFNγ signaling and MHC expression, thus protecting tumor cells. Evaluating Ptpn2 using the CHIME system, Sharpe found that deletion of Ptpn2 in T cells promoted early expansion, induced increased granzyme B production, and provided a competitive advantage over wild-type T cells during LCMV infection. Investigating whether Ptpn2 played a role in controlling subpopulations of exhausted cells, Sharpe found that Ptpn2 deletion led to an increase in terminally exhausted T cells (highly cytotoxic, but less persistent and less proliferative) by releasing the brakes on type I IFN signaling, thus enhancing early differentiation of this population. Ptpn2 deletion did not affect the numbers of progenitor exhausted T cells. In a tumor setting, Ptpn2 deletion in CD8+ T cells improved cytotoxic responses (as it did in the LCMV model), and Ptpn2 deletion in the entire hematopoietic compartment induced total clearance of MC38 tumors, dependent on the presence of CD8+ T cells. Using B16 tumors to evaluate whether this strategy might synergize with PD-1 blockade, Sharpe and colleagues showed that while blockade of PD-1 showed no effect as a monotherapy in this model, it enhanced T cell cytotoxicity and induced notable antitumor effects in mice with Ptpn2 knocked out in hematopoietic cells.
Myeloid Compartment in the TME
Targeting macrophages to promote anti-tumor immunity - Judith A. Varner, UCSD Moores Cancer Center, San Diego, California
Interested in why cytotoxic T cells and NK cells are often excluded from tumors, preventing them from infiltrating and attacking, Judith Varner of the UCSD Moores Cancer Center talked about the contribution of the myeloid compartment and how it can be modified to enhance cancer immunotherapy. With normal functions related to wound healing, macrophages often accumulate in tumors where they support tumor progression through immunosuppression. Prognostic of poor survival, macrophages tend to accumulate rapidly in tumors, where in certain cancers they ultimately outnumber other immune cells. In order to stop myeloid cell-mediated immunosuppression, Varner and colleagues have investigated strategies to block myeloid cell trafficking, prevent proliferation, and repolarize cells towards a less suppressive phenotype. Targeting the recruitment of immunosuppressive macrophages, Varner investigated Ras, PI13γ and downstream effectors, which play roles in activating adhesion molecules, thereby promoting myeloid cell recruitment into tumors. Blocking PI13γ or the upstream chemokine receptor CCR2 suppressed the accumulation of neutrophils and Gr1lo myeloid cells; however, a large population of Gr1- myeloid cells was not affected. Investigating these distinct myeloid cell populations, researchers in Varner’s lab identified Gr1lo myeloid cells as poorly proliferative bone marrow-derived macrophages (BMDMs), and Gr1- myeloid cells as highly proliferative tissue-resident macrophages (TRMs). While BMDMs arrived and accumulated early in murine Lewis lung carcinoma (LLC), they did not express genes associated with inflammation or immunosuppression. TRMs, on the other hand, were highly immunosuppressive and eventually outnumbered BMDMs during tumor progression. In lung cancer patients, the TRM gene signature was associated with poor prognosis. To target the accumulation TRMs, Varner’s lab used cKit inhibitors to suppress macrophage proliferation, inducing slight antitumor effects in mice. To enhance these antitumor effects, Varner and her team turned to 3G8, a novel, high potency cKit inhibitor that simultaneously inhibits several related kinases, including CDK8, which is highly expressed in TRMs. In vitro, 3G8 potently inhibited macrophage proliferation and had modest antitumor effects. In vivo, 3G8 improved immune profiles within tumors (fewer TRM and BMDM; more NK cells, DCs, and T cells), completely eradicated LLC and head and neck cancer, and protected mice from rechallenge. This work shows that targeting macrophages can serve as an effective immunotherapeutic strategy. Further, these strategies could potentially synergize with T cell-targeted therapies to induce strong antitumor effects.
Combination immunotherapy targeting myeloid populations - Charles G. Drake, Columbia University, New York City, New York
Through two narratives, Charles Drake of Columbia University investigated how targeting myeloid cells in the tumor microenvironment (TME) can support immune checkpoint blockade therapy. In the CANTOS Study, a trial of patients surviving myocardial infarctions, antibody blockade of IL-1β prevented secondary cardiovascular events, but also, interestingly, reduced incidence of lung cancer in a dose-dependent manner. Studying this effect in the Renca mouse model of renal cell carcinoma, anti-IL-1β in concert with anti-PD-1 improved therapeutic outcomes compared to either monotherapy, increasing the ratio of M1-type to M2-type macrophages in the TME. Spectral cytometry analysis demonstrated that anti-IL-1β and anti-PD-1 each altered the immune composition of the TME, but in different ways – likely complementary effects, given the treatment’s efficacy. This result has been recapitulated in an ongoing clinical trial of neoadjuvant anti-PD-1 and anti-IL-1β: tumors from treated patients showed a reduction in M2-type and increase in M1-polarized macrophages. Next, Drake considered myeloid targeting in prostate cancer. Following androgen deprivation therapy (ADT) in a mouse model, residual epithelial cells within the TME expressed a variety of myeloid chemoattractants, especially CXCL15 (the mouse analog of IL-8). Additionally, both mouse and human castrate-resistant cancer cells emerging after ADT expressed higher levels of IL-8 at both the transcriptional and protein levels than castration-sensitive cells. In line with these findings, myeloid-derived suppressor cells (MDSCs) dominated the TME of resurgent castrate-resistant cancer. Mechanistically, androgen receptor binding to the IL-8 promoter repressed IL-8 secretion from epithelial cells, which was reversed on ADT or upon preventing androgen receptor signaling. Blocking CXCR2, the receptor for IL-8, prevented infiltration of myeloid-derived suppressor cells (MDSCs) into the TME and enhanced CD4+ and CD8+ infiltration – effects which were reversed as tumors became castration-resistant. In an ongoing clinical trial, prostate cancer patients will be treated with anti-PD-1 with or without anti-IL-8, along with ADT, targeting this myeloid pathway to potentially improve antitumor outcomes.
CAR cell therapy
Clinical development of BCMA-directed therapies in multiple myeloma - Kristen M. Hege, Celgene Corporation, San Francisco, California
B cell Maturation Antigen (BCMA) is a promising target for multiple myeloma (MM), with early encouraging preclinical data and a favorable safety and efficacy readout from a Phase I study with Ide-cel, a 4-1BB, CD3ζ CAR T cell investigational product. Building on these data, Kristen Hege of Bristol Myers Squibb presented one of two keynote talks on day one of the conference. Preclinically, Ide-cel demonstrated low tonic signaling and activity in the presence of circulating soluble BCMA (sBCMA). In the phase I study, analysis of the level of sBCMA early in treatment revealed an inverse correlation with durable response, and rising levels correlated with tumor progression, suggesting that a simple blood test for sBCMA could be a useful biomarker. Moreover, the level of stem cell memory T cells, with LEF1 as a specific biomarker, were a product-intrinsic feature indicative of durable response. In the subsequent phase II pivotal KarMMa trial, Ide-cel continued to demonstrate impressive clinical effects among 140 treated patients, with high, dose-related improvements in response rate, including compete responses (82% overall response rate with 39% complete responses at the highest dose) across multiple patient subgroups. Further, a low incidence of grade 3 or higher cytokine release syndrome (CRS) and neurotoxicity (~6%) was observed, sBCMA levels were confirmed as a biomarker of durable response, and minimal evidence of antigen loss was found in relapsed patients. Comparison of these results to outcomes of a retrospectively collected, stringently evaluated ‘Real World Evidence’ patient cohort confirmed statistically significant clinical improvement with Ide-cel. An improved version of Ide-cel is under development and prepared by T cell growth in the presence of a PI3 kinase inhibitor, which enhances the memory cell phenotype, as evidenced with multiple T cell markers and animal testing. Also in early clinical development is a T cell-engaging, off-the-shelf anti-BCMA product (with two anti-BCMA domains, one anti-CD3 domain, and an immunologically ‘silent’ Fc region). Among 30 patients in a dose-esclating phase I study, most showed low level CRS (grade 1 or 2); one patient had grade 3 CRS and progressed to grade 5. Both objective response and complete response rates were high and dose-dependent, and no antigen loss was observed, which may reflect the importance of BCMA as a survival factor for myeloma cells.
The future of CAR therapies - Elizabeth J. Shpall, The University of Texas MD Anderson Cancer Center, Houston, Texas
Despite the unprecedented therapeutic success of CAR T cell products, particularly in hematologic malignancies, significant toxicities (cytokine release syndrome [CRS] and neurotoxicity) and high costs are areas for improvement. Elizabeth Shpall of Texas MD Anderson Cancer Center outlined approaches for development of similar targeted products from other immune effector cell (IEC) populations, with emphasis on cord blood-derived NK cell products, where an allogeneic, off-the-shelf product could lead to a dramatic decrease in cost and rapid access for patients in need of these life-saving therapies. NK cells are particularly attractive as IECs based on an extensive history of safe use as allogeneic products, with minimal to no graft-versus-host disease and multiple alternative sources (from healthy donors, banked cord blood, induced pluripotent stem cells, cell lines) that are each being engineered with CAR vectors. Moreover, as innate immune cells, NK cells can specifically target tumor cells through their complex set of germline surface receptors. Shpall described her results with cord blood NK cell expansion across multiple donors, which allowed for over 100 doses of CAR-NK cells to be prepared from one unit of cord blood. Moreover, cord blood-derived NK cells typically show higher proliferation and expression of genes involved in cell division than NK cells derived from adult peripheral blood. In a phase I dose-escalating study led by Katy Rezvani, 11 heavily-pretreated patients with chronic lymphocytic leukemia (CLL) were dosed with IL-15-armored CD19 CAR-NK cells derived from cord blood, with no CRS or neurotoxicity, including 2 patients who had only 1-2 of 6 HLA match (other patients were 4/6 HLA-matched). Seven complete responses were observed, including one in more advanced Richter’s stage CLL. The CAR-NK cells persisted for up to 12 months – an unexpected result, as allogeneic NK cells are typically rejected, though this might be due to stimulation through the CAR.
Next generation CAR T cells for cancer therapy - Renier J. Brentjens, Memorial Sloan Kettering Cancer Center, New York, New York
In his talk on next generation CAR T cells, Renier Brentjens of Memorial Sloan Kettering Cancer Center described current CAR T cells as the “Ford Model T”, when what we really need is a “Ford Mustang”. In other words, while current CAR T cells are the product of incredible progress, there is much work to be done to improve, refine, and optimize this form of cancer treatment. In order to expand the applications of CAR T cells and enhance responses, several strategies are being pursued, including enhancing cytotoxicity and persistence, using CAR T cells to recruit the endogenous immune system, using CAR T cells to target the immunosuppressive TME, and enhancing the safety profile of CAR T cells. One way to improve CAR T cells is to enhance the design of the receptor itself. Current second and third generation CARs utilize signaling mechanisms that mimic TCR stimulation (signal 1) and costimulation (signal 2) in natural T cells. In recent studies, researchers have found that altering the number of functioning ITAM domains or changing a single residue in CD28-signaling domain of CARs could reprogram T cell effector and memory functions in a way that favors CAR T cell persistence, inducing long-term remissions in mice. Brentjens also described several forms of “armored CAR T cells”, which express CARs in combination with other factors that could potentially improve functionality. For example, CAR T cells modified to express costimulatory ligands on their surface (like 4-1BB or CD40 ligands) could enhance T cell activation, while CAR T cells modified to express cytokines (like IL-18 or IL-36γ) could enhance activation, epitope spreading, and safety profiles. Armored CAR T cells can even be engineered to secrete PD-1-blocking scFvs, which could mimic the effects of checkpoint inhibitors. Thus far, each of these strategies has been shown to significantly enhance antitumor efficacy and/or long-term survival in mice. With these, and many other potential enhancements to CAR T cell therapies on the horizon, “Model T” CAR T cells will likely be getting some major upgrades in the near future.
NK cells
Innate immune defenses against cancer: Potential for mobilizing NK cells for cancer immunotherapy - David H. Raulet, University of California Berkeley, Berkeley, California
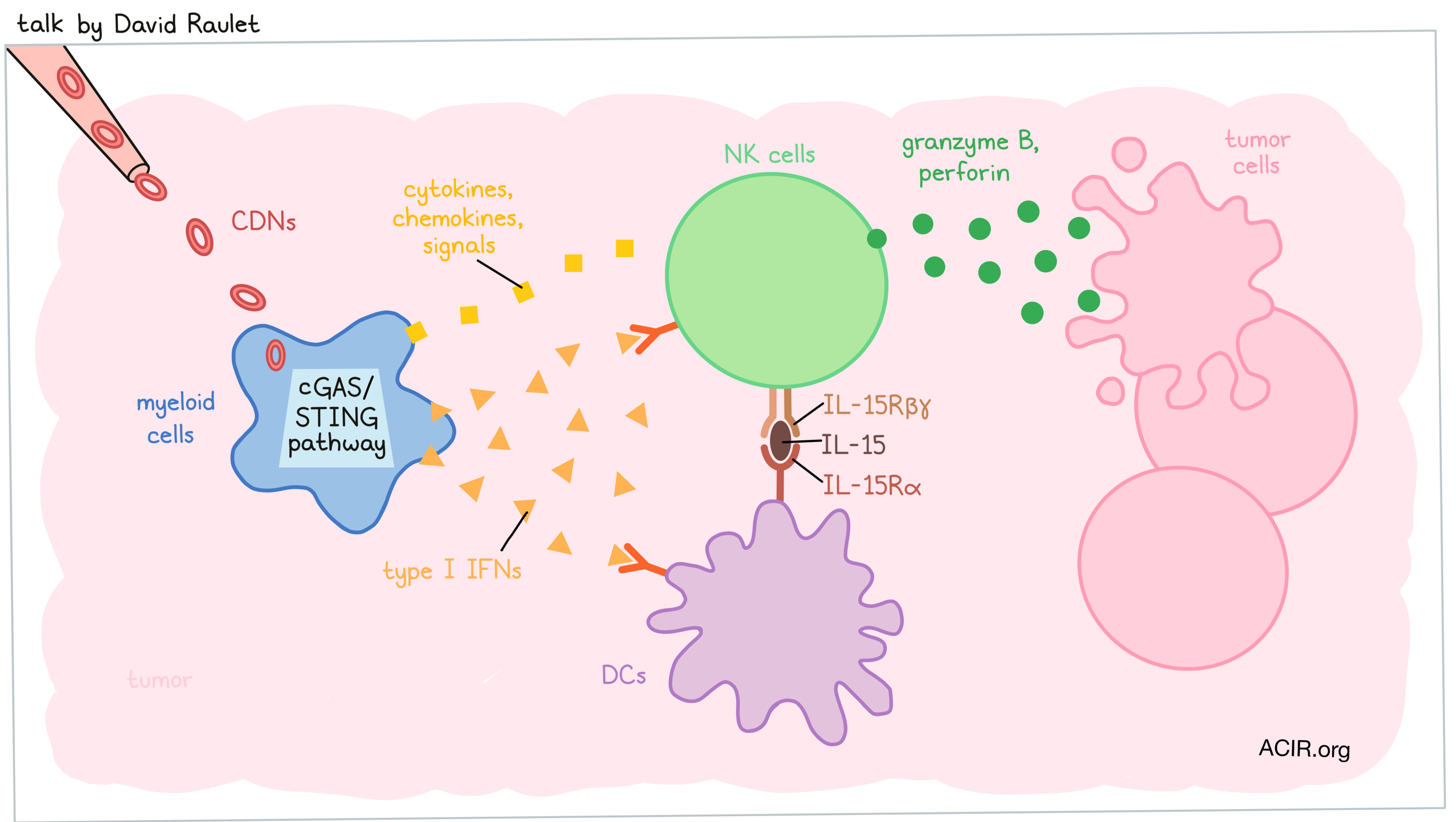
While the majority of cancer immunotherapies focus on T cells, David Raulet of the University of California Berkeley is focused on the underdog, seeing huge potential in mobilizing NK cells. Much like T cells, NK cells are capable of directly killing tumor cells, but unlike T cells, they are not dependent on antigen availability or MHC expression, and in fact thrive in environments in which MHC is downregulated. NK cells respond to stress-induced activating ligands, which are broadly expressed in tumors. A major mechanism for the spontaneous mobilization of antitumor NK cell activity is activation of the cGAS-STING pathway, which Raulet’s group has previously shown can act in different cells. When cytosolic DNA, which is often constitutively present in cancer cells, activates this pathway and cyclic dinucleotides are transferred to myeloid cells, downstream transcription factors can activate an innate immune response in myeloid cells, which, in turn, promote and support NK cell (and T cell) responses. This pathway can be supercharged by injecting cyclic dinucleotides (CDNs) directly into tumors. In mice bearing MHC-I-deficient tumors that were resistant to T cell-mediated antitumor effects, intratumoral administration of CDNs served as an effective NK cell-mediated immunotherapy. This strategy was effective in a number of tumor models, inducing cures and long-term survival in many mice. Further investigation revealed that NK cells accumulated in tumors treated with CDNs and were systemically activated. To determine whether this systemic NK cell activation might induce antitumor effects at distant tumor sites, Raulet’s team injected Rag2-/- mice with dual-flanked tumors and treated one side. Treated tumors were profoundly rejected and contralateral tumors also showed evidence of antitumor effects, suggesting that NK cells had some systemic antitumor efficacy, but did not provide complete systemic protection. Investigating the underlying mechanisms of NK cell responses following CDN therapy, Raulet and colleagues found that the effect was dependent on type I IFN activation of both NK cells and DCs. In DCs, IFN-induced activation was found to induce IL-15/15Rα complexes on the surface, and blocking IL-15/15Rα abrogated tumor rejection. This led Raulet to propose a model in which CDNs act primarily on myeloid cells, which produce type I IFN, activating both NK cells and DCs. In response to IFN, DCs upregulate IL-15/15Rα, which further enhances NK cell responses. Other cytokines, chemokines, and signals also seem to play a role. Testing these effects in NK-sensitive MHC-I+ tumors in Rag2-/- mice, Raulet and colleagues found that CDNs induced tumor rejection that was at least partially mediated by NK cells. Testing possible combination therapies involving CDNs, the researchers found that IL-2 family cytokines, particularly a stabilized IL-2 “superkine”, super2-MSA, synergized with CDNs by enhancing NK cell activation. The combination of CDNs and super2-MSA was highly effective in several mouse models, and even showed antitumor efficacy in the notoriously difficult-to-treat Kras/p53 model of primary sarcoma.
The IRE1 ER stress sensor activates natural killer cell immunity by regulating c-Myc - Laurie H. Glimcher, Dana-Farber Cancer Institute, Boston, Massachusetts
In the keynote presentation for day two of the conference, Laurie Glimcher of the Dana Farber Cancer Institute described her recent work in the decades-long journey studying the important endoplasmic reticulum (ER) stress-induced signalling pathway involving the ER1α unfolded protein sensor and its target, the xbp1 transcription factor (which is activated by a novel mechanism wherein the ribonuclease domain of ER1α is released upon activation, transits to the nucleus, and removes a 26 nt segment of the pre-xbp1 RNA, resulting in the functional xbp1 protein). Past work has shown this pathway is essential for the development of plasma cells from B cells and the development of other professional secretory cell types (such as pancreatic acinar cells). Given the multiple stresses in the cancer tumor microenvironment, her work has also shown how activation of this pathway in multiple immune cell types leads to dysfunction in dendritic cells and impaired metabolism in T cells, and how knockout of the pathway improves tumor control. Now turning to the role of NK cells, a highly secretory (IFNγ, granzyme, perforin) cell type that shows a proliferative burst in response to triggers, resulting in innate memory, Glimcher showed that the ER1α/xbp1 pathway is essential for viral control, as well as the proliferative burst. Moreover, in the B16 melanoma model, the pathway was essential for controlling metastasis to the lung, mechanistically related to decreased NK cell infiltration in the lung and subsequently reduced cDC and T cell recruitment. The ER1α/xbp1 pathway is also essential to NK cell reconstitution by homeostatic expansion following lymphodepleting treatments, supporting the use of inducers of ER1α, such as IL-12, IL-15, and IL-18, to pre-treat NK cells being prepared for adoptive therapy. Finally, Glimcher and her team discovered that the ER1α/xbp1 pathway also controls the transcription of the key regulatory molecule MYC, with MYC deletion phenocopying xbp1 deletion, and overexpression of MYC overcoming xbp1 defects. Overall, these results highlight how the ER1α/xbp1 pathway has opposite, but important roles in different cell types of the immune system, and suggests approaches to intervene in a cell-type specific way to enhance tumor and viral control.
by Lauren Hitchings, Alex Najibi, and Ed Fritsch



