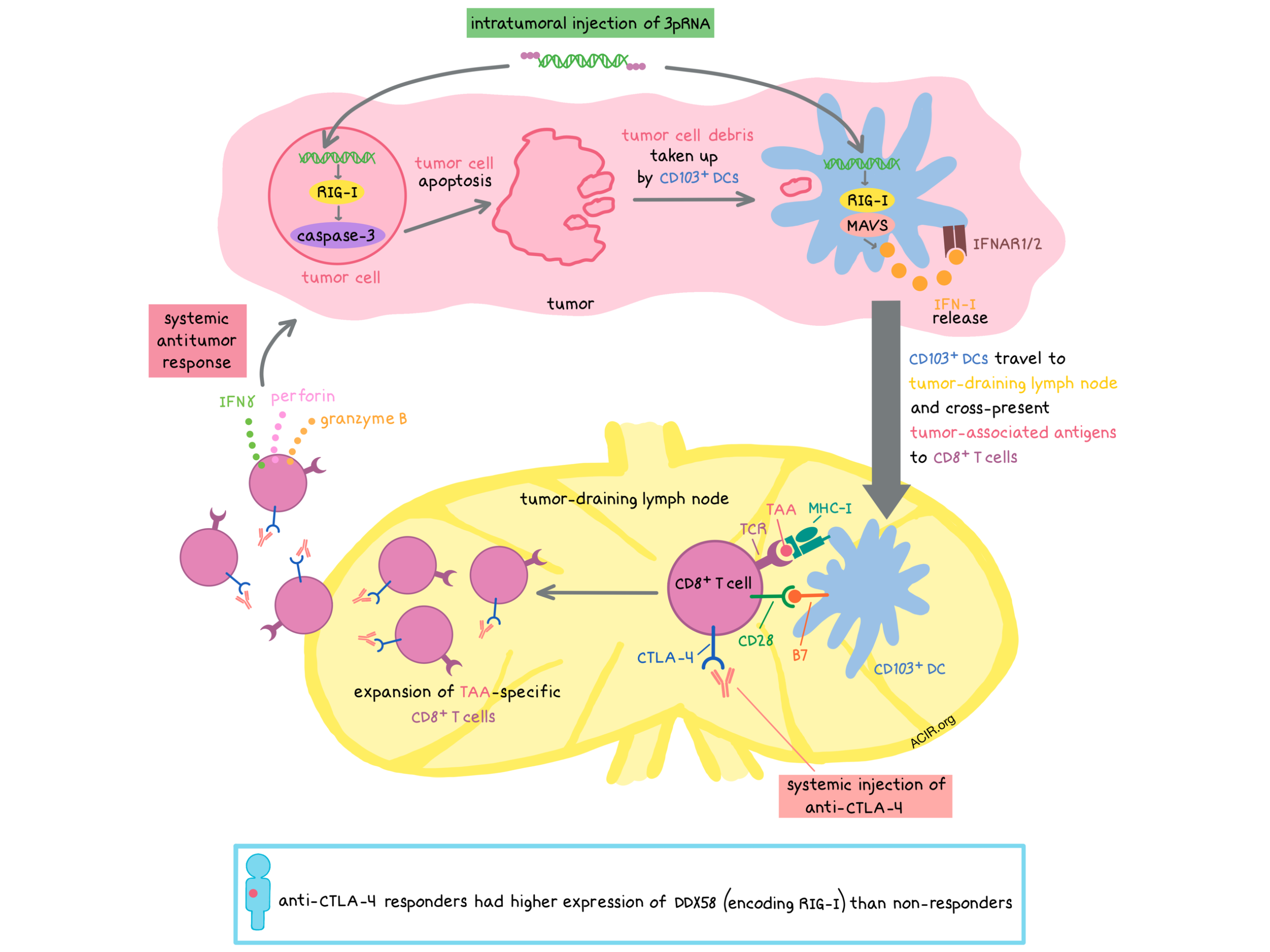
Aiming to improve the consistency of response to checkpoint inhibitors, Heidegger and Wintges et al. explored how the signaling of RIG-I – a cytosolic sensor that detects double-stranded RNA during bacterial or viral infection – contributes to IFN-I production within the tumor microenvironment, and how it affects the antitumor immunity mediated by immune checkpoint blockade. The results, recently published in Science Immunology, demonstrate a critical role for RIG-I signaling in tumor cells in both mice and humans.
The researchers began by utilizing the immunogenic OVA-expressing B16 melanoma cell line (B16.OVA) in vivo. Mice were subcutaneously implanted with either wild-type (WT) B16.OVA or RIG-I-/- B16.OVA tumors, and were treated intraperitoneally with either anti-CTLA-4 or control antibodies. Anti-CTLA-4 slowed tumor growth in both WT and RIG-I-/- models, but much less so in the latter, resulting in reduced survival. These results indicate that tumor cell-intrinsic RIG-I signaling plays a role in anti-CTLA-4-mediated immune response.
To test whether therapeutically targeting RIG-I would enhance the response to anti-CTLA-4, the researchers implanted either WT or RIG-I-/- B16.OVA tumors into the right and left flanks of mice (with the same tumor genotype in each side), injected 3pRNA (double-stranded 5’-triphosphate RNA which activates RIG-I) into the tumors in the right flank, and treated some of the mice intraperitoneally with anti-CTLA-4. 3pRNA injection alone completely eradicated the injected (“local”) WT tumors, but not the non-injected (“distant”) WT tumors. Combination of 3pRNA and anti-CTLA-4 controlled both local and distant WT tumors and promoted long-term survival and immunological memory in most treated mice. RIG-I-/- local tumors were not as well controlled with 3pRNA injection as WT tumors. Combination of 3pRNA and anti-CTLA-4 did not reduce growth of distant RIG-I-/- tumors, resulting in reduced survival compared with mice bearing WT tumors. In mice with tumors of different genotypes in the two flanks, the tumor injected with 3pRNA had to be WT in order to mediate regression of the distant tumor. Together, these experiments demonstrate that local activation of tumor-intrinsic RIG-I enhanced the systemic antitumor response to anti-CTLA-4.
Next, Heidegger and Wintges et al. explored the mechanisms behind the effect of RIG-I signaling on anti-CTLA-4 antitumor response. The researchers observed in vitro and in vivo that tumor cell-intrinsic RIG-I signaling induced caspase-3-mediated apoptosis of tumor cells and (in vitro) that tumor cell debris was taken up via phagocytosis by dendritic cells (DCs). CD103+ DCs cross-presented tumor-associated antigens in the tumor-draining lymph nodes (TDLNs), and this cross-presentation was enhanced by anti-CTLA-4 and/or 3pRNA treatment, leading to systemic expansion of OVA-specific cytotoxic T lymphocytes (CTLs) in a manner dependent on RIG-I signaling. The largest increase in CTL expansion and infiltration into injected and distant WT tumors was observed with combination treatment.
Furthermore, utilizing Mavs-/- mice (which lack the essential RIG-I adapter molecule MAVS) inoculated with WT tumors, the researchers demonstrated that host RIG-I signaling was required for systemic expansion of tumor-specific CD8+ T cells and for control of distant tumors (but not local tumors) following treatment with 3pRNA and/or anti-CTLA-4 therapy. Overall, these results demonstrate that in the B16.OVA tumor model, tumor cell-intrinsic RIG-I signaling was required for the control of local (injected) tumors while host RIG-I signaling was required for the control of distant (non-injected) tumors following combination 3pRNA/anti-CTLA-4 therapy. In addition, Mavs-/- mice bearing RIG-I-/- tumors completely failed to generate an antitumor response to any treatment, indicating that RIG-I signaling, either in tumors or in host, is necessary for the efficacy of anti-CTLA-4 therapy. In addition, looking at the double-stranded DNA-sensing cGAS/STING pathway, which is known to play a role in anti-CTLA-4 efficacy, the researchers interestingly found that it is the host, but not tumor cell, STING signaling that was required for anti-CTLA-4-mediated antitumor response in the B16.OVA model.
Knowing that RIG-I ligation induces IFN-I production in myeloid cells, leading to innate and adaptive immune responses, Heidegger and Wintges et al. explored whether RIG-I signaling in tumor cells produced a similar effect. RNAseq analysis of bulk tumors revealed that RIG-I signaling was associated with cytokine production (IFNα, IFNγ), apoptosis, and inflammatory signaling. Furthermore, Heidegger and Wintges et al. showed that IFN-I signaling was required for the antitumor effects of anti-CTLA-4. However, it was the host, and not the tumor cell, production of IFN-I that, together with tumor cell-intrinsic RIG-I signaling, promoted anti-CTLA-4 antitumor efficacy.
In the poorly immunogenic B16-F10 melanoma model, anti-CTLA-4 monotherapy was ineffective, but the combination of anti-CTLA-4 and 3pRNA induced a systemic antitumor response that led to rejection of the local tumors and delay in the growth of distant tumors, resulting in prolonged survival. The antitumor response was due to the systemic expansion of endogenous CD8+ T cells reactive against the melanoma-associated antigen TRP2. A similar benefit was observed in the pancreatic Panc02 model. In contrast, such synergy was not observed in the 4T1 mammary carcinoma model. The researchers found that 4T1 cells were resistant to RIG-I-mediated apoptosis.
Turning to PD-1 blockade, the researchers showed that anti-PD-1 reduced tumor growth and prolonged survival in mice bearing WT or RIG-I-/- B16.OVA tumors, although to a lesser extent than anti-CTLA-4. However, the anti-PD-1-mediated antitumor effect was not dependent on tumor-intrinsic RIG-I signaling. Combination of anti-CTLA-4 and anti-PD-1 elicited a strong antitumor response in mice with WT or RIG-I-/- B16.OVA tumors and prolonged their survival, with intact tumor-intrinsic RIG-I signaling conferring a larger benefit.
Finally, seeking to confirm the clinical relevance of these findings, the researchers analyzed RNAseq TCGA data. In a cohort of 456 patients with primary melanoma, high expression of DDX58 (encoding RIG-I), but not TMEM173 (encoding STING), was associated with longer overall survival, while low DDX58 expression was shown to be an independent risk factor for death. In a small cohort of 18 patients with melanoma being treated with anti-CTLA-4, RNAseq data revealed that responders had higher DDX58 expression than non-responders. In a cohort of 51 patients with metastatic melanoma who had been treated with anti-PD-1, tumor RNAseq analysis revealed no correlation between high expression of DDX58 and overall survival.
Overall, the preclinical and clinical data support the conclusion that high RIG-I transcriptional activity correlates with longer overall survival and durable response to anti-CTLA-4. These findings suggest that testing human tumor biopsies for RIG-I signaling and/or expression may help predict outcomes of anti-CTLA-4 treatment, and that clinically targeting RIG-I signaling may be beneficial.
by Anna Scherer
Meet the researcher
This week, lead author Hendrik Poeck answered our questions.

What prompted you to tackle this research question?
The unresponsiveness of our own tumor patients to checkpoint blockade and our longstanding expertise in innate immune signaling that made us connect these pathways.
What was the most surprising finding of this study for you?
The two most surprising findings were that the TCGA data of metastatic melanoma patients identified low RIG-I expression as an independent risk factor for death and that high tumoral RIG-I-encoding DDX58 expression may serve as a potential biomarker for ICB efficacy in the future.
What was the coolest thing you’ve learned (about) recently outside of work?
That Corsica, a mountainous Mediterranean island, is the perfect spot for an active family vacation as it has the perfect mixture of mountain sports and beach at every level.



