In the early stages of tumor development, the process of autophagy (degradation of intracellular components) protects against the emergence of malignant cells; however, in more established cancers, autophagy can promote tumor progression by supplying the tumor cells with necessary nutrients. Inspired by this duality, Cunha et al. explored non-canonical roles of autophagy components and found that one such alternative function, LC3-associated phagocytosis (LAP) within tumor-associated macrophages (TAMs), contributes to immune suppression within the tumor microenvironment (TME). The results were recently published in Cell.
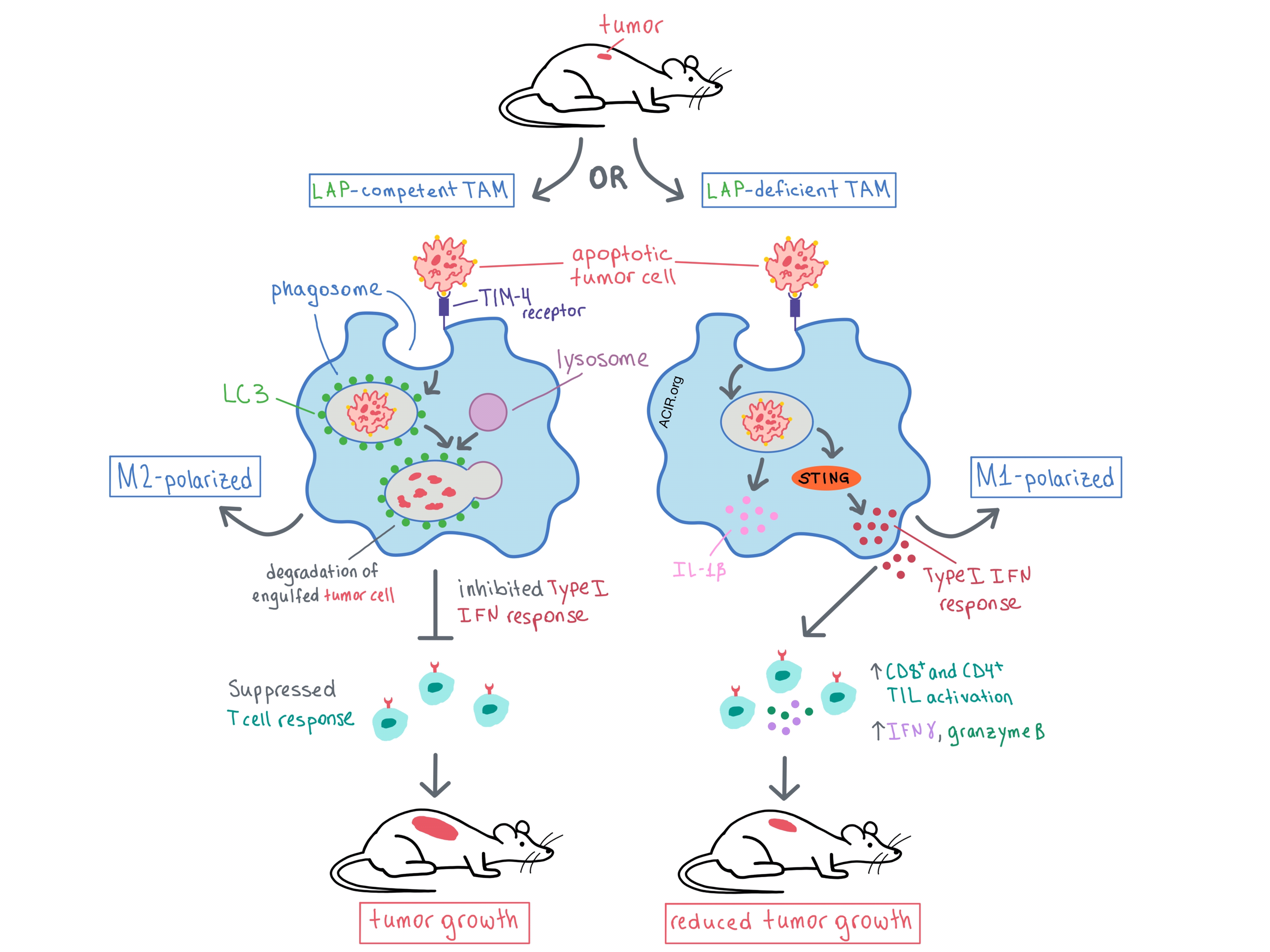
LAP is triggered by engagement of Toll-like receptors, Fc receptors, or TIM-4 (the apoptotic cell receptor) on the phagosome during the engulfment of dead or living cells. Some of the components of the autophagy machinery (including microtubule-associated protein 1 light chain 3 [LC3], which is typically found on autophagosomes) are recruited to the phagosome, leading to its maturation, fusion with a lysosome, and eventual degradation of the engulfed material. However, beyond its role in inducing cargo degradation, LAP has been found to occur in several types of innate immune cells, and has been shown to regulate macrophage immune response. Therefore, the researchers utilized selective genetic ablation to tease out the relevant components and roles of LAP in TAMs and other myeloid cells within the TME.
Previously, the team had demonstrated that some autophagy proteins are involved in both canonical autophagy and LAP (BECN1, VPS34, ATG5, ATG7, ATG16L1), while others affect only autophagy (FIP200, ULK1, ATG14) or only LAP (RUBCN, NOX2). With this knowledge, the researchers used lysozyme M (LysM/Lyz2)-Cre-lox recombination to selectively ablate specific genes (and therefore autophagy and/or LAP) in monocytes, macrophages, and CD11b+ dendritic cells and observed the effect on tumor growth. Tumor growth was suppressed in immunocompetent, LAP-deficient mice with subcutaneously engrafted B16F10 melanoma, Lewis lung carcinoma (LLC), or MC38 colon adenocarcinoma, but no tumor suppression was observed when genes necessary only for canonical autophagy were ablated, although the canonical autophagy process itself was compromised. Similar effects were observed in a genetic model of non-small-cell lung cancer (NSCLC). Together, these results indicated that in myeloid cells, LAP, but not canonical autophagy, promotes tumor growth.
The researchers then analyzed mechanistically how LAP affects different cell compartments. They found that LAP deficiency had no effect on the number of infiltrating myeloid cells, granulocytic PMN-MDSCs, or monocytic Mo-MDSCs. However, LAP deficiency reduced the surface expression of CD206 on macrophages, indicating reduced M2 polarization. The authors concluded that LAP leads to polarization of TAMs toward the immunosuppressive M2 phenotype.
Digging deeper into the effect of LAP in TAMs, Cunha et al. performed single-cell RNAseq and gene expression analysis on non-granulocytic cells obtained from LLC-engrafted mice that were either LAP-proficient or LAP-deficient. The analysis identified a cluster of LAP-deficient, mature TAMs (Ly-6Clo MHC-IIhi) with decreased expression of the M2 markers Mrc1 and Arginase-1, further supporting the role of LAP in M2 polarization. In another cluster of LAP-deficient, monocytic TAMs (Ly-6Chi MHC-IIlo), there was an increased expression of type I IFN target genes. Following implantation of a fluorescently labeled LLC cell line, the researchers showed that upon phagocytosis of a dying tumor cell, LAP-deficient TAMs increased their expression of type I IFN target genes, as well as IL-1β and IFNβ genes. Similar results were observed in vitro in LAP-deficient, bone marrow-derived macrophages. These data suggested that LAP in TAMs inhibited IFN production following engulfment of apoptotic tumor cells, and that the increase in IFN response in LAP-deficient mice likely led to the reduced tumor growth observed.
Next, the team explored the mechanism behind the antitumor response that is observed with LAP deficiency. First, via deletion of the IFNα/β receptor IFNAR in mice, the researchers confirmed that the type I IFN response was indeed necessary for the antitumor effect of LAP deficiency. Although phagocytosed dying cells contain DNA, the cytosolic DNA sensor STING, which can upregulate the type I IFN response, is typically not triggered. However, Cunha et al. observed that in macrophages deficient in LAP, but not in both LAP and STING, the type I IFN pathway was upregulated, indicating that LAP prevents the activation of STING when apoptotic cells are engulfed. Extending this observation, in LAP-deficient mice the antitumor effect and the polarization of TAMs toward the M1 phenotype was abrogated when STING was deleted. Closing the mechanistic analysis, the researchers found that it was the STING-dependent type I IFN response that led to increased CD8+ and CD4+ tumor-infiltrating lymphocyte (TIL) activation and IFNγ production in LAP-deficient mice. Depletion experiments demonstrated that the antitumor effect in LAP-deficient mice was dependent on both CD8+ and CD4+ TILs.
Overall, Cunha et al. have shown that phagocytosis of apoptotic tumor cells triggers LAP to polarize TAMs toward the M2 phenotype, leading to suppressed TIL function and, ultimately, tumor growth. Deficiency in LAP polarizes TAMs toward the proinflammatory M1 phenotype and triggers a STING-dependent type I IFN response, which promotes TIL activation and production of IFNγ, reducing tumor growth. These results suggest that targeting the LAP pathway may have therapeutic potential.
by Anna Scherer



