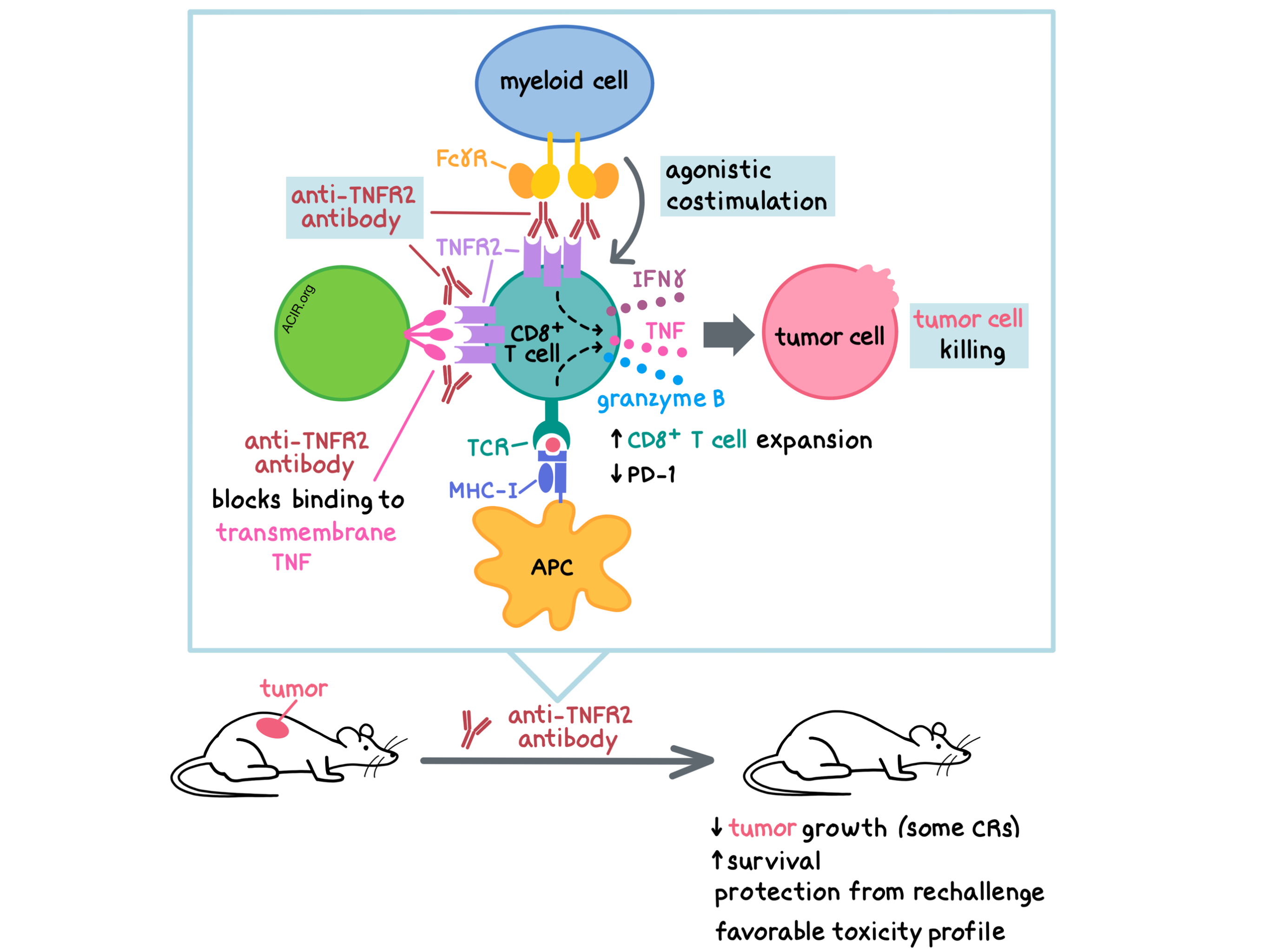
Searching for new cancer immunotherapies that are effective and well tolerated, Tam, Fulton, and Sampson et al. explored targeting tumor necrosis factor receptor 2 (TNFR2), which is mostly expressed on immune cells, including Tregs and activated T cells. In a paper recently published in Science Translational Medicine, the researchers generated and characterized anti-murine and anti-human TNFR2 antibodies that exert antitumor effects via Fc-dependent agonism.
The researchers began by generating a panel of mouse IgG2a anti-TNFR2 antibodies and evaluating them in syngeneic tumor models. Epitope mapping allowed the researchers to identify five novel anti-TNFR2 antibodies that bound to unique epitopes covering all four cysteine-rich domains (CRDs) of the murine TNFR2. Three of the five antibodies blocked TNF binding and, together with TCR stimulation, induced the proliferation of CD8+ T cells in vitro. One of the antibodies, Y9 (targeting CRD1 and blocking TNF binding), activated both CD8+ and CD4+ T cells (including Tregs) in vitro and upregulated the expression of granzyme B, CD25, and PD-1 on CD8+ T cells, suggesting that it was an agonist.
In mice with established CT26 colorectal tumors, all five anti-TNFR2 antibodies reduced tumor growth and prolonged survival, with Y9 conferring the largest benefit. Since two of the antibodies reduced tumor growth without blocking TNF binding, the researchers suspected that blocking TNF binding was not necessary for the antitumor response. Given these results, Tam, Fulton, and Sampson et al. examined the antitumor effect of Y9 in several additional tumor models. They found responses in seven models, including six with some complete responses (CT26 colorectal carcinoma, EMT6 mammary carcinoma, WEHI-164 fibrosarcoma, MC-38 colon adenocarcinoma, anti-PD-1-sensitive Sa1/N bladder sarcoma, and anti-PD-1-resistant MBT-2 bladder cancer) and one with only partial responses (A20 B lymphocyte sarcoma). In contrast, 4T1 mammary carcinoma, B16-F10 melanoma, and LLC1 Lewis lung carcinoma did not respond to treatment. Combination of Y9 and anti-PD-1 or anti-PD-L1 synergized in the CT26 and EMT6 models to prolong survival. Furthermore, in CT26, EMT6, or WEHI-164 tumor models, Y9 induced immunological memory and protected mice from rechallenge.
To investigate the mechanism of action of Y9, Tam, Fulton, and Sampson et al. began by examining the role of Fc binding in the antitumor response. To do this, they generated Y9-DANA, a variant of Y9 with Fc mutations that abrogate binding to Fcγ receptors (FcγRs), and demonstrated in three tumor models that FcγR binding was required for the antitumor activity of Y9. Since Y9 and Y9-DANA had comparable blocking of TNF binding, but Y9-DANA lacked antitumor efficacy, the researchers concluded that blocking the TNF/TNFR2 signaling axis was not the main mechanism of Y9. Using mice with a knockout of either the inhibitory Fc receptor (FcγRIIb) or the activating receptors (FcγRI, FcγRIII, and FcγRIV), the researchers showed that both the activating and the inhibitory FcγRs contributed to the antitumor activity of Y9. To evaluate the relative contributions of the activating and inhibitory FcγRs, the researchers created Y9 variants with various IgG isotypes that had different affinities for the activating and inhibitory FcγRs and tested their activity in vivo. The Y9 variants with an increased affinity for the inhibitory FcγRIIb did not lose antitumor activity compared with the original Y9 antibody, suggesting that the main mechanism of action of Y9 in vivo is enhanced agonism mediated by FcγR clustering. However, the contribution of other potential mechanisms, such as antibody-dependent cellular cytotoxicity or antibody-dependent cellular phagocytosis (both mediated by the activating FcγRs), could not be ruled out.
Next, the researchers analyzed the immune cell types involved in the Y9-mediated antitumor activity. Depletion experiments showed that Y9 activity was mediated by CD8+ T cells and NK cells, but not CD4+ T cells. Y9 did not appear to directly act on tumor cells. Examining the immune cells within the tumors and tumor-draining lymph nodes, the researchers observed decreased surface expression of TNFR2 after Y9 treatment on CD8+ T cells, conventional CD4+ T cells, and Tregs. At the same time, levels of soluble TNFR2 in the serum increased. Decreased surface level expression and increased soluble levels of TNFR can occur due to shedding of the TNF receptors after clustering-induced signaling; thus, these observations further support the hypothesis that the Y9 antibody acts by inducing costimulatory signaling. Y9 treatment led to the expansion and increased functionality (increased IFNγ and TNF production, decreased PD-1 expression) of tumor antigen-specific CD8+ T cells. Increased numbers of IFNγ+CD8+ T cells correlated with reduced tumor mass. Y9 treatment did not deplete Tregs in vivo. Together, these data suggest that the main mechanism of Y9 is costimulation of CD8+ T cells via TNFR2 agonism.
Toxicity profiling of healthy and tumor-bearing mice undergoing long-term treatment with Y9 revealed no weight loss, splenomegaly, liver damage, or histological changes in lymphoid or nonlymphoid tissues. Y9 (either alone or in combination with anti-PD-1) did not cause spontaneous activation or proliferation of peripheral T cells, and did not upregulate the costimulatory CD86 molecule on DCs. Y9 treatment only moderately and transiently increased serum TNF and IL-6 levels. Overall, the Y9 anti-TNFR2 antibody exhibited a favorable toxicity profile, particularly in comparison with the IgG-matched anti-CTLA-4.
Finally, aiming to develop an anti-human TNFR2 antibody with properties similar to the murine Y9 antibody, Tam, Fulton, and Sampson et al. generated two antibodies: Ab1, a chimeric antibody derived from mouse immunization, and Ab2, a human antibody derived from a human scFv phage library. Both Ab1 and Ab2 targeted the CRD1 region of the human TNFR2 without competing with each other for TNFR2 binding. Similar to Y9, both Ab1 and Ab2 blocked the binding of TNF to TNFR2. Ab1 and Y9 were found to bind structurally equivalent epitopes on the human and mouse TNFR2, respectively (data for Ab2 could not be obtained), suggesting that the mouse data could be highly translatable to the clinic. In vitro, Ab1 and Ab2 increased the proliferation, cytokine levels, and activation markers in CD8+ and CD4+ T cells from the peripheral blood of healthy human donors. In several humanized mouse models, Ab1 and Ab2 slowed tumor growth, either as monotherapies or in combination with anti-PD-1. Combination treatments were more efficacious than anti-PD-1 alone.
Overall, these results demonstrate that the anti-human TNFR2 antibodies induced costimulation of T cells in vitro and mediated antitumor responses in vivo, supporting the development of agonistic anti-TNFR2 antibodies in the clinic.
by Anna Scherer
Meet the researcher
This week, Eric Tam and Andreas Raue answered our questions together.

What prompted you to tackle this research question?
ET and AR: It has long been recognized that the mechanism of action of antibodies targeting the TNF superfamily of receptors can be quite complicated. For GITR, OX-40, and 4-1BB – some of the more well-known members of this receptor family – the relative contributions of agonistic activity, blocking of the activity of the natural ligand, and depletion of regulatory T cells (Tregs) have been extensively discussed only after the first drug candidates had entered clinical trials, potentially leading to less optimal outcomes. When we started to work on TNFR2, a relatively less well-described target for cancer immunotherapy, we were surprised by how little attention this topic had received. Prompted by some initial data that contradicted the prevalent opinion at the time – that blocking TNF should be the desired mechanism for antibodies targeting TNFR2 – we decided to investigate the mechanism of the most active antibodies we found in our screens in an unbiased way.
What was the most surprising finding of this study for you?
ET and AR: As experimental evidence started to accumulate that agonistic activity rather than ligand blocking was driving the activity in syngeneic mouse tumor models, the team was at first puzzled how this can be reconciled with research from autoimmune and transplant literature that was suggesting that agonistic activity would lead to an enhancement of the suppressive capacity of Tregs. The assumption to date was that if the TNFR2 agonism would have a positive outcome in autoimmune disease, then the opposite effect (TNFR2 antagonism/blocking of signaling) should be the best strategy for cancer immunotherapy. We continued to let the experimental evidence guide our research and in the end, our studies showed that the antibody that had the greatest antitumor activity was agonizing the TNFR2 receptor both in vitro and in animal tumor models.
What was the coolest thing you’ve learned (about) recently outside of work?
AR: Over the summer, I finally realized that the temperature of the ocean in New England is not always necessarily freezing cold. There seem to be very rare, but favorable, conditions where air temperature, wind, tide, and the local beach geometry can result in water temperature that is just tolerably warm. Unfortunately, the data points I collected so far are too sparse to make predictions. Therefore, most of the time we’d already spent the 25 bucks for the parking before we find out if we can go in more than ankle deep. Another big unknown in this equation is whether big whites would find the warmer spots also more appealing, something I hope I will never find out more about.



