
Last week, the ACIR team attended the Fifth CRI-CIMT-EATI-AACR International Cancer Immunotherapy conference in Paris, France. This week’s extensive special feature covers select talks from the conference, which are organized by the topics below.
Targeting the PD-1 axis
Vaccination approaches
CARs targeting solid tumors or the tumor stroma
Tumor and the microenvironment
Microbiota-immune interactions
Biology of innate immune cells and therapeutic approaches
Targeting the PD-1 axis
Ton Schumacher of the Netherlands Cancer Institute opened CICON 2019 by presenting a novel approach to understanding the immunological response to PD-1 axis blockade in human cancers. His Tumor Fragment (TF) platform consisted of newly resected tumor fragments that were carefully divided into multiple samples and stored. Cell recovery and antibody penetration studies indicated that such samples were suitable for ex vivo analysis and perturbation. 37 tumor samples from multiple histologies, including primary and metastatic lesions, were treated ex vivo with anti-PD-1 antibodies and the impact on T cell activation and cytokine/chemokine production was evaluated. Clustering revealed a clear impact in ~⅓ of the samples (TF-Responders), with the most pronounced effects being in upregulated CCL20, CXCL1, and IL-2. LCK inhibition linked this effect to TCR signaling, and IFNγR blockade showed IFNγ dependence. Of the 11 patients in the study who had been subsequently treated with anti-PD-1 therapy, clinical responders demonstrated TF ex vivo response to anti-PD-1 while clinical non-responders did not. Deeper analysis of cell composition by flow cytometry, spatial distribution by immunohistochemistry, and steady-state cytokine/chemokine production ultimately revealed 3 classes of TF non-responsive tumors (immune desert; T cell-excluded; and T cell-infiltrated but immunologically quiescent) and a TF responder class with T cell infiltration and high baseline production of cytokines/chemokines, particularly CXCL13. Building on prior work associating tumor-infiltrating T cell phenotypes (CD39+/ CD103+ and PD-1T [referring to very high PD-1 expression]) with clinical response to PD-1 blockade, the presence of PD-1T cells and CXCL13 production, particularly their localization in tertiary lymphoid structures, were the best predictors for anti-PD-1 responsiveness and hence potential biomarkers for a clinical effect.
Ira Mellman of Genentech presented evidence and arguments challenging the current dogma that anti-PD-1 axis therapy works or solely works by reinvigorating exhausted T cells in the TME. Multiple hints supported the rationale for this challenge: dysfunctional markers are also activation markers; no hard evidence exists to show that exhausted cells are dysfunctional; blocking the PD-1 axis in vitro does not enhance cell killing; PD-1 signaling may enhance T cell longevity and reduce activation-induced cell death. Moreover, recent published work demonstrating a critical role for CD28 in PD-1 signaling, and some recent unpublished work demonstrating that the PD-L1/B7.1 interaction only occurs in cis, and data suggesting that the PD-1/PD-L1 interaction is prevented by B7.1 sequestration of PD-L1 together have indicated that a 1:1 relationship (i.e. interactions and signaling) between PD-1 and PDL-1 or B7.1 and CD28 was oversimplified. Clinical IHC data has pointed to an important role of myeloid cell-expressed PD-L1 in the TME and Mellman described experiments demonstrating that the critical PD-L1-expressing cells were CLEC9A cross-presenting dendritic cells (despite the predominant expression of PD-L1 by CD11b+/F480+ macrophages in the TME). Given the role of DCs in T cell priming, Mellman presented data showing enhanced vaccine efficacy in PD-L1 knockout mice. Finally, evaluation by single-cell sequencing of TCR clonotypes in tumor, adjacent normal tissue samples, and peripheral blood from patients being treated with atezolizumab showed that T cell clonotypes expanded in tumor and adjacent normal tissue were also found in peripheral blood, had an effector/effector memory phenotype, and correlated with clinical response. Overall these results point to PD-1 axis blockade working at the level of T cell priming in tumor-draining lymph nodes to enhance T cell proliferation and replenish tumor-resident T cells, and suggest that the focus for improvements should be on T stem cell memory enhancements, such as vaccines, cytokines, or dendritic cell stimulators.
Vaccination approaches
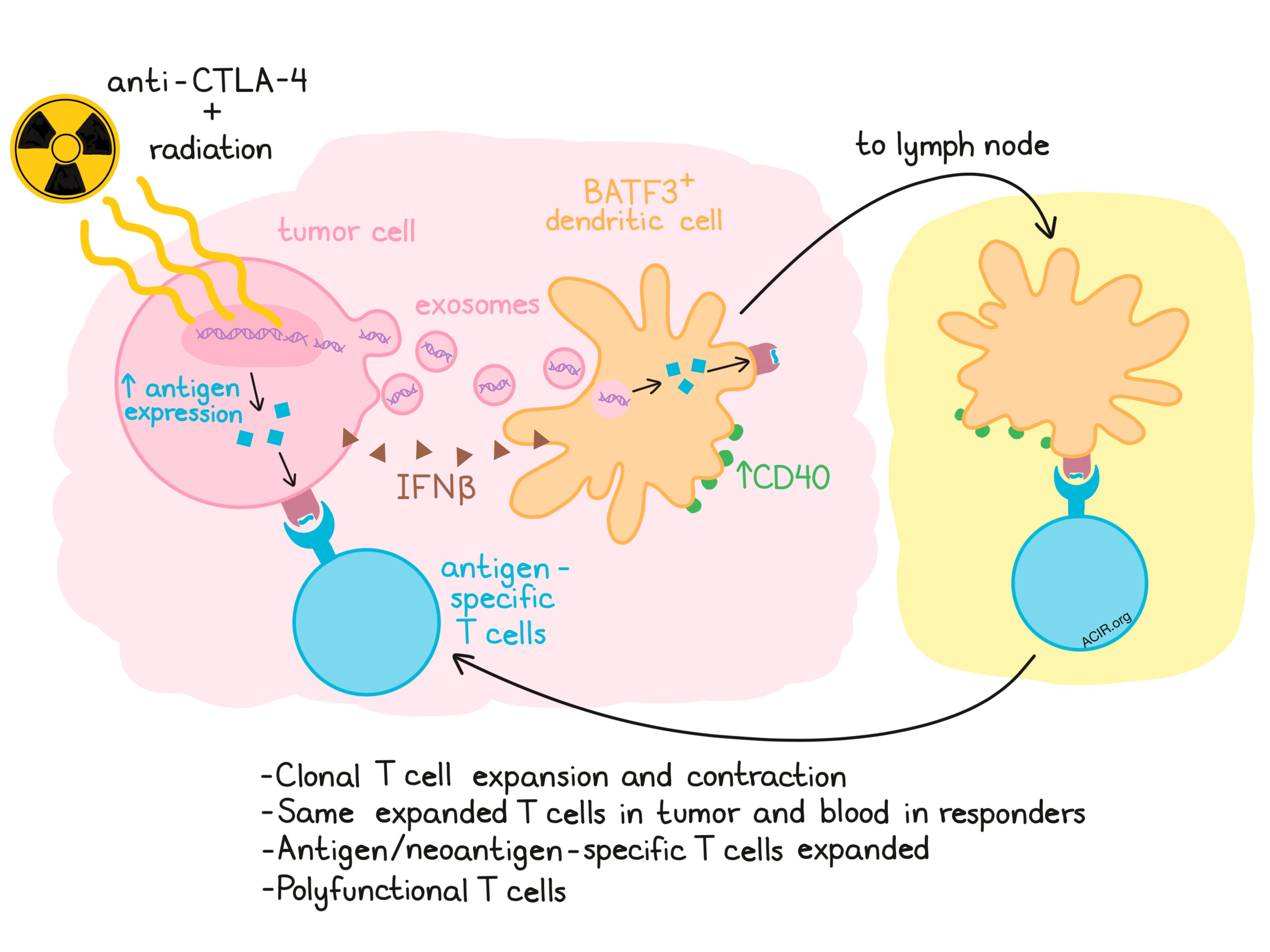
Sandra Demaria of Weill Cornell Medical College evaluated the mechanism of in situ vaccination radiation therapy, discussing preclinical and clinical results in conjunction with anti-CTLA-4 blockade. Using the mouse 4T1 model, which contains the retroviral dominant antigen AH-1 and is classically considered a “cold” tumor, Demaria showed that targeted radiation coupled with anti-CTLA-4 led to a significant enhancement of AH-1-specific CD8+ T cells in the tumor-draining lymph node while either therapy alone was comparable to background. Radiation-induced DNA damage and release activated the cGAS-STING pathway and IFNβ production in the tumor cell, mimicking an antiviral response. Demaria was able to show that the tumor DNA was delivered to dendritic cells (DCs) via exosomes and that Batf3+ DCs increased in number and showed enhanced cell surface expression of CD40. This led to a cGAS-dependent increase in IFNγ in the lymph node. CD8+ T cells were increased in an abscopal tumor. In a recent clinical trial of radiation combined with anti-CTLA-4, which showed 2 CRs and 5 PRs among 39 chemorefractory patients with non-small cell lung cancer, circulating INFβ levels 3 weeks after one dose of anti-CTLA-4 and one cycle of radiation tracked with clinical response. Only patients who experienced a CR or PR showed expansion or contraction of individual TCR clonotypes at 3 weeks in the peripheral blood. A subset of the expanded clones were also detected in the tumor prior to treatment and expanded in the tumor during treatment. Nucleic acid sequencing and peptide-MHC binding predictions were used to determine potential immunogenic neoantigens for one patient, resulting in the detection of T cells reactive to two different epitopes of a mutated antigen (KPNA2) after treatment. One epitope was a de novo response, the other was boosting of a pre-existing response. Radiation enhanced the expression of KPNA2. Returning to the 4T1 mouse model to conduct a more in-depth analysis of responding T cells, Demaria showed that the clonality of intratumoral T cells increased after combination treatment and that this was primarily driven by radiation. Single-cell RNAseq revealed large numbers of CD8+ and CD4+ T cell clusters and a diversity of transcriptional states for AH1-specific CD8+ T cells, which differed based on whether mice received radiation, anti-CTLA-4, neither, or both treatments. A gene expression signature derived from two of these clusters was predictive of survival in a clinical breast cancer trial.
Özlem Türeci of Biontech presented initial clinical results for Melanoma FixVac, a liposomal RNA vaccine (RNA-LPX) targeted at 4 non-mutated tumor-associated antigens (TAAs) expressed in a large proportion of melanoma patients. The TAA targets are the well-known antigens tyrosinase, MAGE-A3, NY-ESO-1, and TPTE, an immunogenic cancer testis antigen found by the group in melanoma and other tumors. RNA-LPX was shown to target to CD11c+ dendritic cells in the spleens of mice. PET/CT scans after initial intravenous injection of the vaccine in humans showed enhanced fluoro-glucose uptake in the spleen, suggestive of similar targeting in humans. Trial eligibility included advanced stage disease (Stage IIIB-C, IV; ~½ of the patients had radiological evaluable disease at the time of treatment). The trial included a dose-escalation phase followed by expansion cohorts, and monitored safety, T cell responses, and clinical response. Some patients also received concomitant anti-PD-1 therapy. In the dose-escalation phase of the trial, dose-dependent increases in a number of inflammatory cytokines (IFN, IL-6, IP-10, IL-12p70) and patient body temperature were observed to spike 2-4 hours after injection and return to baseline within 24 hours. ELISPOT analysis demonstrated de novo induction of responses to at least one TAA in more that 75% of patients and expansion of pre-existing responses in almost all patients. Both CD4+ and CD8+ T cell responses were observed and immunogenic epitopes for multiple HLA alleles were detected. Tetramer staining demonstrated that some responses showed TCR polyclonality, and induced T cells were shown to be cytotoxic. Most CD8+ T cells were effector memory cells that produced cytokines upon antigen stimulation. Clinical outcomes were reported for 42 patients with measurable disease, all but one of whom had prior checkpoint therapy. Of the 25 patients treated with FixVac alone, 16% showed a partial response and 28% had stable disease. Of the 17 patients treated with FixVac and anti-PD-1, 35% showed a partial response and 12% had stable disease. Examples of patients who had progressed on prior checkpoint therapy but developed partial responses on FixVac or FixVac + anti-PD-1 were shown, including a patient who may have been sensitized by vaccination to subsequent anti-PD-1 therapy.
CARs targeting solid tumors or the tumor stroma
Ellen Puré from the University of Pennsylvania addressed targeting tumor stroma, particularly cancer-associated fibroblasts (CAFs), to undercut this essential support for tumor cells. Fibroblasts undergo a “stromagenic switch” when they transition from quiescent, tumor-inhibitory cells to activated cells which actively support the maintenance and growth of tumor cells and suppress antitumor immunity. Eliminating or reprogramming CAFs would become an orthogonal approach to tumor control. There are multiple CAF subsets but survival correlations in multiple cancers, including pancreatic cancer, highlight two main CAF subsets: (1) the fibroblast activation protein (FAP+) subset, which is considered the inflammatory/activated subset that needs to be removed and (2) the smooth muscle actin (SMA+) subset, which is tumor-restraining. FAP+ CAFs are found in multiple human tumor types and in murine tumors. There are different approaches to removing FAP+ CAFs and Puré focused on the development of a FAP-directed CAR T cell. Using an OVA-expressing mouse model, Puré showed that FAP+ CAF cell depletion led to disruption of desmoplastic stroma, reduced angiogenesis, and reduced tumor growth; the latter was dependent on host FAP. In addition, FAP-directed CAR T cell treatment increased CD8+ T cell infiltration and the functionality of the intratumoral T cells, and a synergy with a vaccine was demonstrated (in a different tumor model), establishing FAP+ CAFs as an immune checkpoint. Alternate approaches to removing FAP+ CAFs include modulating the regulatory circuits (calcineurin-NFAT; cytokines; TGFβ) or structural regulators (proteins and matrix stiffness/elasticity).
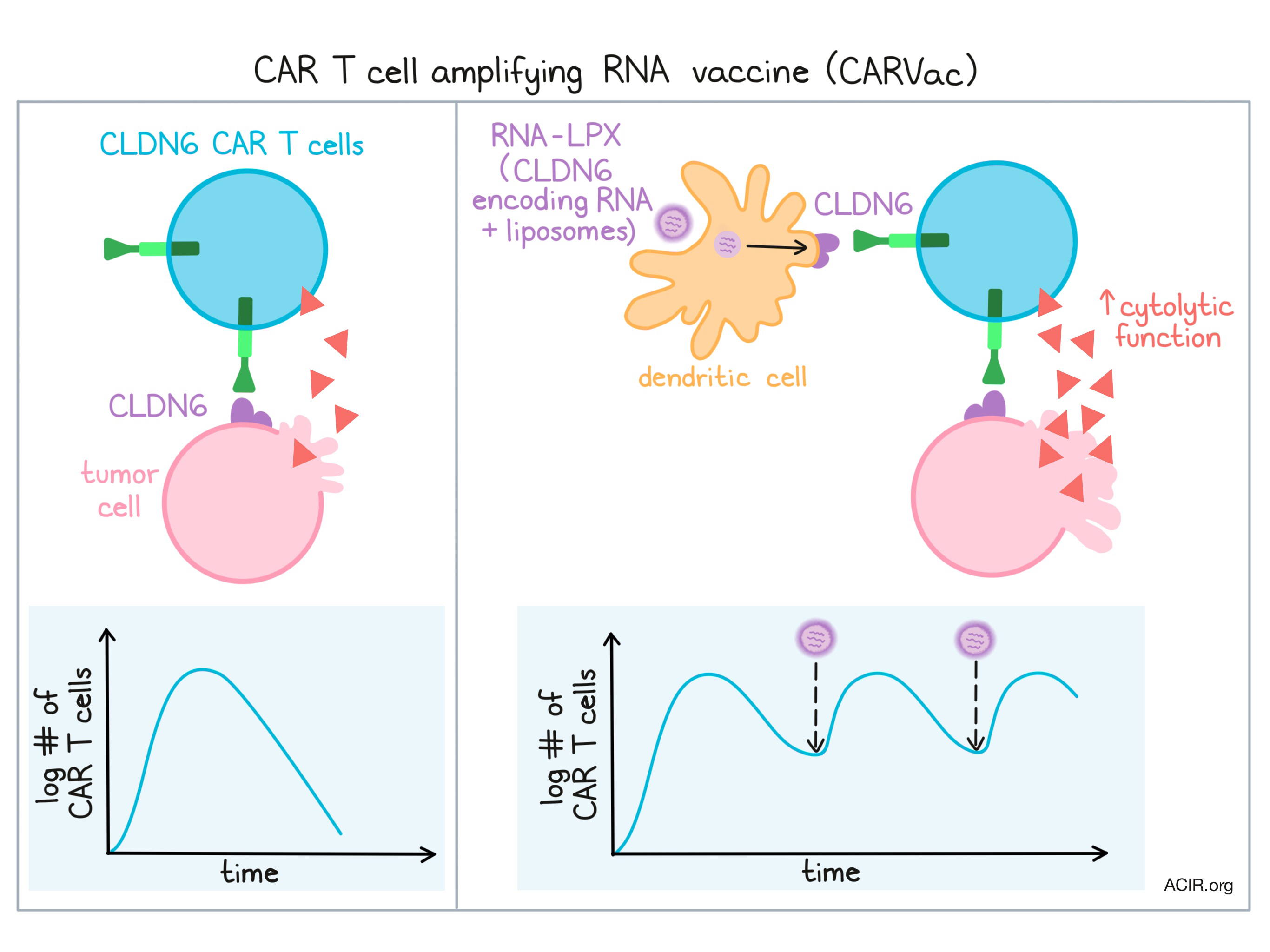
Katharina Reinhard of Biontech presented the preclinical work supporting CARVax, an RNA vaccine intended to amplify antigen-specific CAR T cells targeting solid tumors. Reinhard began by describing the rationale for choice of claudin6 as a CAR target. Claudin6 is a developmentally regulated gene expressed in many cancer types and is a cancer stem cell marker. In vitro and in vivo experiments demonstrated that the scFv-CD8 hinge-4-1BB-CD3ζ claudin6-targeting CAR construct showed specific binding, and CAR-transduced T cells were cytolytic in tumor spheroid assays (even repetitively cytolytic following addition of a second spheroid after tumor cells in the first spheroid were killed) and completely cleared tumors in a mouse xenograft model. Concerned that the persistence of solid tumor-targeting CAR T cells would be limited by a need for antigen stimulation and the tissue-sequestered nature of solid tumors compared to blood cancers, Reinhard additionally developed a liposomal RNA (RNA-LPX) claudin6 vaccine for antigen-presenting cell (APC)-targeted delivery. Claudin6 is a membrane protein, and by expressing full-length claudin6 in the vaccine, the expectation was that APCs taking up the RNA-LPX would express claudin6 on their surface and serve to re-stimulate the CAR T cells even after the bulk of the initial tumor was removed. In vitro and in vivo experiments confirmed this expectation by demonstrating that repetitive vaccination maintained significant levels of the claudin6 CAR T cells. Effector T cells were initially observed but eventually effector memory and central memory cells developed. In vivo, vaccine-stimulated claudin6 CAR T cells showed excellent tumor cell clearance (even when starting with a low number of CAR T cells) and superior ex vivo properties (IFNγ and TNF production and cytotoxicity).
Tumor and the microenvironment
Thea Tlsty from the University of California at San Francisco outlined the downstream effects of epithelial cell stress on other cell types, which she has been studying for years in multiple contexts, and concluded by identifying loss of CD36 expression as a key step leading to a desmoplastic, pro-tumor stroma. Cross-talk between fibroblast types (normal or cancer-associated fibroblasts) and normal or tumorigenic epithelial cells led to dramatic tumor growth when the tumorigenic forms of both were implanted in mice. Analysis of RNA and protein expression in breast tissues of low- and high-mammographic density demonstrated that the fibroblast expression of CD36 was dramatically reduced in the higher density samples. Loss of fibroblast expression of CD36 was both necessary and sufficient to induce the desmoplastic state, led to increased angiogenesis, and promoted tumor progression. Loss of CD36 is observed across multiple tumor types and subtypes, and may occur independently of (and not as a consequence of) tumor cell signals. Interestingly, CD36 on macrophages is necessary for phagocytosis of phosphatidylserine-containing apoptotic cells, but pro-tumor M2 macrophage dramatically downregulate CD36 expression, preventing clearance of dead cells, stimulating inflammation and ultimately tumorigenesis. Adenosine released from tumor cells appears to result in the downregulation of CD36 on macrophages.
Investigating the mechanisms for tumor regrowth after initial response, Sjoerd van der Burg of Leiden University set up a TC-1 model in which suboptimal E7 synthetic long peptide vaccination results in reproducible initial tumor elimination followed by regrowth. Neither the quantity nor quality (phenotype, cytokine secretion) of target-specific T cells in the blood or spleen were different at the time of initial response or tumor regrowth. Furthermore, similar high percentages of E7-specific T cells expressed checkpoints (PD-1, LAG3, TIM3) at both initial regression and regrowth. Treatment with various checkpoint inhibitors (anti-PD-1, anti-PD-L1, anti-NKG2A) did not prevent regrowth (although it slightly delayed regrowth) and attempts to enhance the vaccine booster dose with agonist antibodies and checkpoint blockade failed. Turning attention to tumor-intrinsic mechanisms, neither tumor antigen expression, surface HLA expression, nor differential sensitivity of the tumor to cytotoxic T cells revealed any differences. Examining the composition of regressing versus relapsing tumors showed a dramatic shift toward immunologically “cold” tumors prior to relapse, showing intratumoral loss of CD8+ T cells and E7-specific T cells with no change in Tregs and an increase in CD11b+ myeloid cells. CD8+ T cells also showed reduced proliferation, perhaps due to myeloid cell inhibition. However, treatment with a CSF1R inhibitor did not prevent relapse. Analyzing the expression profile of relapsed tumors revealed high TGFβ production, but TGFβ blockade did not improve relapse. To prove the tumor cell-intrinsic nature of the resistance, relapsed tumors were re-implanted into naïve mice and showed immediate resistance. Interestingly, the re-implanted tumors were well infiltrated with CD8+ T cells but were deficient in inflammatory myeloid cells (Ly6Chi, CD11b+). Cisplatin, which induces inflammation and restores inflammatory myeloid cell infiltration in TC-1 tumors, prevented regression of tumors in the suboptimally vaccinated mice.
Given the importance of DC1 (BATF3+CD103+) dendritic cells to tumor control and response to immunotherapy, Barbara Maier from the Icahn School of Medicine at Mount Sinai Hospital studied dendritic cells within the mouse KP lung tumors with the hope of improving their number or functionality. Single-cell RNAseq revealed not only the expected DC1 and DC2 populations, but also a new population of DC1s with a distinct transcriptional profile characterized by upregulated expression of regulatory molecules such as PD-L1 and PD-L2, SOCS1 and SOCS2, and CD80/86; upregulation of molecules involved in migration; and downregulation of TLRs. These DCs were named mregDCs. mregDCs were enriched in tumor-bearing compared to naive animals. Using GFP as an antigen (so that antigen-ingesting cells in lung tumors could be followed), Maier found that the mregDC state was induced in GFP+ DC1s, confirmed by an enhanced mregDC signature in GFP+ cells and by flow cytometry. Interestingly, AXL, a receptor found on tumor-associated macrophages, was upregulated on mregDCs and was required for upregulation of PD-L1. Induced mregDCs also upregulated genes involved in a Th2 response, including IL4Rα, which can lead to reduced IL-12 production following exposure to IL-4 in vitro. IL-4 blockade, but not PD-L1 blockade, enhanced GFP-specific CD8+ T cell proliferation and function in vivo, leading to significantly reduced tumor growth. Presumably, within the tumor-bearing lung, factors driving a Th2 immune response, such as IL-4, may be responsible for induction of the mregDC1 phenotype, reducing effective Th1 immunity and providing an opportunity for improvement.
Microbiota-immune interactions
Chengcheng Jin of the Massachusetts Institute of Technology explored the role of the microbiota in lung tumors spontaneously developing in KP mice under germ-free (GF) versus specific pathogen-free (SPF) conditions. Jin examined tumor infiltration by microbes and the resulting immune cell signature. GF mice developed fewer, smaller, less proliferative, and lower grade tumors than their SPF counterparts – an observation that was mimicked in SPF mice treated with antibiotics. Analysis of lungs of SPF mice with and without tumors demonstrated differences in both the extent of microbial bioburden (higher in tumor-bearing lungs) and which bacterial strains were present. Moreover, there was a clear association with tumor and microbial bioburden in the lung, but no correlation in the spleen. Intratracheal inoculation of microbes from lungs of tumor-bearing mice into naive KP SPF mice accelerated spontaneous tumor formation. Concomitant changes in the immune cell populations were examined by flow cytometry and demonstrated a dramatic increase in γδT cells in tumor-bearing lungs with minor but significant increases in the blood and spleen. The transcription factor RORγT and cytokine IL-17, required for γδT cell differentiations, were both specifically upregulated in newly tumor-infiltrating cells. Antibody blockade of the γδTCR (Vγ6, Vδ1) or of activating IL-17 reduced tumor growth, pointing to a pro-tumorigenic effect of γδT cells. γδT cells infiltrating tumor-bearing lungs were distinct from those infiltrating the spleen, specifically upregulating IL-22 (an epithelial cell growth factor) and amphiregulin (an EGFR ligand). CD11b+Ly6G+ neutrophils also accumulated in tumor-bearing SPF lungs, apparently in response to γδT cells – as γδTCR or IL-17 blockade reduced neutrophil infiltration. Analysis of TCGA data revealed 1) bacterial dysbiosis compared to normal lung, 2) γδT cell increases in human lung cancer tissues, and 3) a correlation between worse survival and the γδT cell gene expression signature found in KP mice. These data point to a vicious cycle of stimulation of microbial infiltration by newly formed tumors, γδT cell infiltration, and production of factors which stimulate further tumor growth. This approach provides a reductionist system to further evaluate these initiation events and the influence of therapeutic interventions.
Building on prior work on the role of Enterococcus hirae in the antitumoral efficacy of cyclophosphamide and checkpoint blockade, Laurence Zitvogel of the Gustave Roussy Cancer Center looked for immunogenic responses to E. hirae in a mouse MCA205 fibrosarcoma model. Mice pretreated with antibiotics showed improved response to cyclophosphamide treatment when they were reconstituted with E. hirae. Noting that not all E. hirae strains were effective, Zitvogel demonstrated that strain 13144 was immunogenic and protective. Epitope analysis revealed that peptides from the tail protein of a lysogenic bacteriophage found in strain 13144 were immunogenic. Tetramer analysis demonstrated that CD8+ T cells specific for this antigen could be observed in the spleen and draining lymph nodes, and that prophylactic vaccination with a single epitope peptide or E. coli expressing the epitope could elicit the protective effect. A search for homologous sequences in the mouse genome, which could inform cross-reactivity of the E. hirae epitope, identified a region of the mouse oncogene PSMB4 that was identical in 7 of 9 positions, including the anchor residues. Genetic knock-in of altered PSMB4 sequences into the MCA205 tumor line abrogated the anti-cancer efficacy of E. hirae, but had no or a minor effect on in vitro tumor growth, pointing to an in vivo immune mechanism. TC-1 lung tumors, which express high levels of PSMB4, also responded to E.hirae, while MC38 colorectal tumors, which express low levels of PSMB4, did not. Clinical data indicated a correlation between positive clinical response and bacteria infected by a bacteriophage with the tail protein, prompting a similar search for homologous human proteins to phage tail protein. The search revealed several HLA-A02:01 epitopes which were immunogenic, one of which had nearly 80% homology to the tumor suppressor glycerol-3-phosphate dehydrogenase, expression of which can correlate with progression-free survival following checkpoint therapy. The phage can become lytic and transfer to other enterococcal species, suggesting the possibility of “phagotherapy”.
Biology of innate immune cells and therapeutic approaches
Building the underlying biology to add a new cell type influencing the immune system, Pavel Hanc of Harvard Medical School (von Adrian lab), described how nociceptors, cells of the afferent nervous system which secrete neuropeptides in response to noxious stimuli (cold, heat, chemicals, pain), interact with dendritic cells (DCs) and influence their function. Early work showed the close anatomical location of nociceptor ganglia and dendritic cells in the skin and that the significant skin inflammation sometimes caused by imiquimod, an approved immunotherapeutic, is ablated when nociceptors are deleted. Using in vitro coculture of bone marrow-derived DCs and isolated primary neurons, Hanc was able to show that the presence of neurons improved production of the key DC cytokines IL-12 and IL-6 upon imiquimod, flu virus, or yeast stimulation. Physical contact between the neurons and DCs was required based on transwell assays; DCs migrated toward neurons via a CCL2 gradient. DCs and neurons formed tight interactions and temporal firing of neurons led to near simultaneous Ca2+ flux within the DCs, suggesting a possible synergistic addition to suboptimal DC stimulation. Both cDC1 and cDC2 were impacted, and a similar enhancement of DC response by nociceptors to imiquimod stimulation could be observed in vivo. Single-cell RNAseq revealed a significant number of genes differentially expressed in multiple pathways (hypoxia, glycolysis, protein secretion), the most significant of which was IL-1β, following coculture with nociceptors and capsaicin stimulation). IL-1β production was directly induced with a series of neuropeptides, and was lost when nociceptors were ablated in mice. This foundational work sets the stage for future investigations.
Eric Vivier of the Centre d’immunologie de Marseille-Luminy bridged the innate and adaptive immune systems with his description of monalizumab, an IgG4 antibody that blocks NKG2A, which together with CD94 forms a heterodimeric, inhibitory receptor found on NK and T cells. NKG2A/CD94 binds HLA-E, which is more ubiquitously and highly expressed than PD-L1 in human cancers. In the A20 lymphoma model, intratumoral NK and T cells express NKG2A, and a large fraction of intratumoral T cells express NKG2A and PD-L1. Blockade of both targets results in improved tumor control in this model, supporting the ongoing clinical study combining monalizumab with durvalumab (anti-PD-L1) in multiple solid tumors. NK cells are capable of ADCC through the activating receptor CD16 (FcγRIII), raising the possibility that blocking the inhibitory NKG2A receptor would potentiate the ADCC activity of NK cells mediated by tumor-targeting antibodies. In vitro studies demonstrated impressive synergy of monalizumab and therapeutic antibodies targeting either EGFR or CD20, which led to a phase II study in head and neck squamous cell carcinoma (HNSCC) combining monalizumab and cetuximab (anti-EGFR). Among 40 patients with a median follow-up of 17 months, the combination was shown to be acceptably safe with an encouraging overall best response rate of 27.5% and overall survival rate of 44% at 12 months. Responses observed in platinum-resistant patients were independent of HPV status and irrespective of prior immunotherapy. Monalizumab thus is a multi-potent antibody working through multiple cytolytic cells (NK and T cells) and multiple mechanisms (checkpoint blockade and ADCC).
by Ed Fritsch



