PD-1 and other known markers of activation and exhaustion have helped researchers to understand T cell biology and develop numerous immunotherapies. While many of these markers are well known and well studied, there are likely more that have yet to be investigated. Looking into the relationship between PD-1 and other markers of activation, Palmer and Webber et al. noticed something interesting about the expression patterns of CISH (cytokine-induced SH2 protein) and did a deep dive into understanding the biology of what could be an important internal checkpoint regulating T cell reactivity. A pre-print version of their findings, which have not yet been peer reviewed, was recently published on bioRxIV.
While studying markers of activation and exhaustion on fresh TIL from treatment-naive melanoma patients, Palmer and Webber et al. noticed that cells expressing activation markers such as PD-1, 4-1BB, TIM3, TOX, and CD39 were distinct from cells expressing CISH. In fact, PD-1 and CISH appeared to be almost mutually exclusive. Further, while CISH+CD8+ T cells were fairly uncommon in peripheral blood samples, they were enriched in tumors. Looking at naive T cells, central memory T cells, and effector memory T cells, the researchers found that each expressed low levels of CISH before TCR stimulation and upregulated CISH significantly following TCR stimulation. This effect was more pronounced with advancing differentiation status, suggesting that CISH expression is induced by TCR stimulation and increases with chronic antigen stimulation.
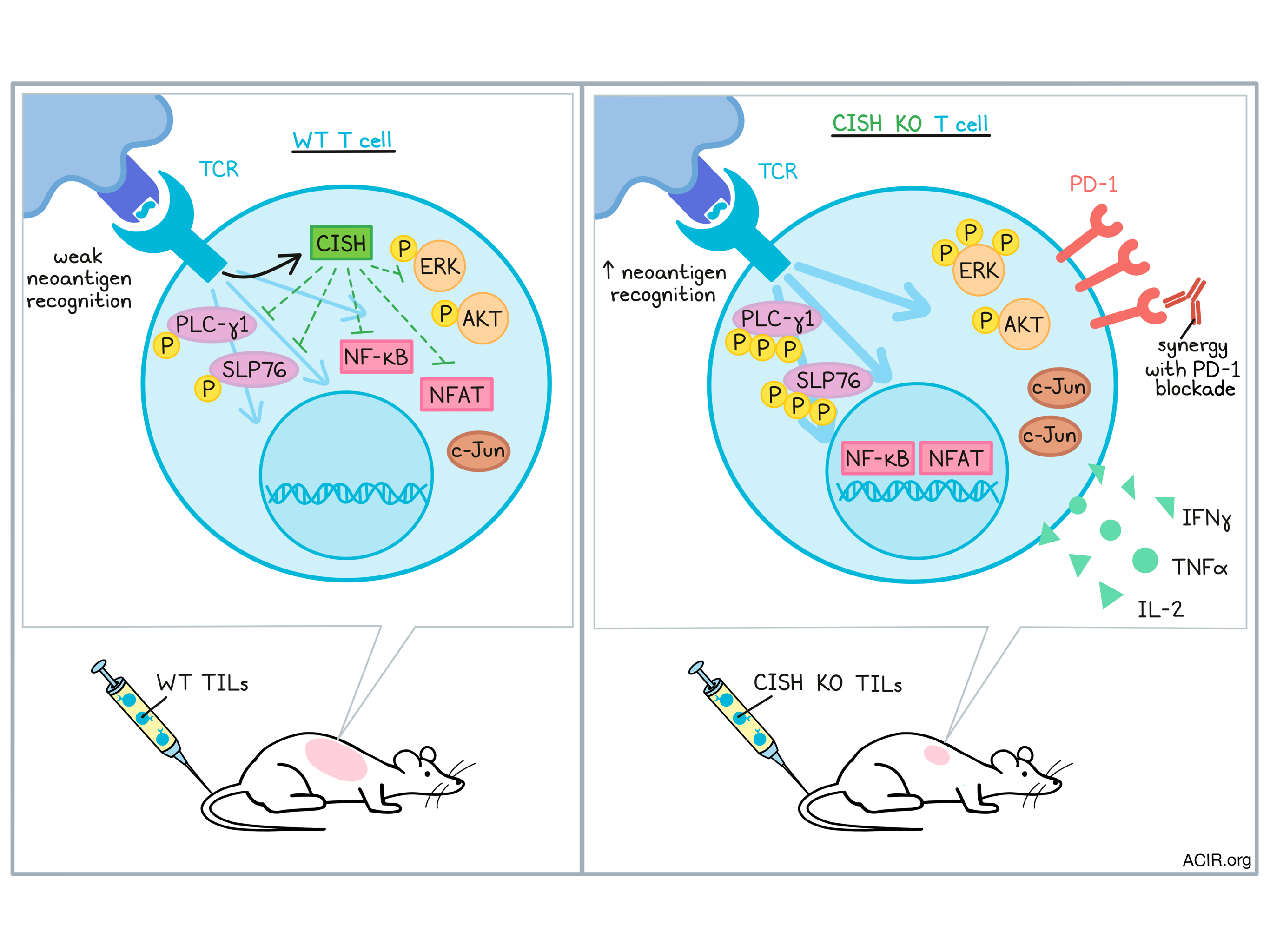
To investigate the functional significance of CISH expression, the researchers developed a CRISPR/Cas9-based strategy to knock out expression of CISH in human T cells. Upon anti-CD3-mediated TCR stimulation, CISH knockout (KO) cells showed increased production of IFNγ, TNFα, and IL-2, and a higher portion of CISH KO cells were polyfunctional compared to wild-type (WT) cells. These results suggest that CISH KO enhanced functional activation after TCR stimulation.
Next, Palmer and Webber et al. engineered CISH KO T cells with an HLA-A2-restricted TCR specific for NY-ESO-1 to evaluate the role of CISH in antigen responses. In coculture with NY-ESO-1+ cells, a higher portion of CISH KO compared to WT cells were polyfunctional – an increase not observed in coculture with NY-ESO-1- cells. This pattern was maintained when CISH KO cells were deployed in an assay against relevant and irrelevant tumors, suggesting that CISH inhibits TCR stimulation in an antigen-specific manner.
Based on this evidence, the researchers hypothesized that CISH KO may be able to restore weak or suppressed T cell responses to neoantigens by enhancing their reactivity to neoantigen-driven TCR stimulation. To study this, they developed a reagent-sparing strategy that allowed them to knock out CISH in TILs prior to ex vivo rapid expansion (REP). The team utilized samples from a patient with metastatic colon cancer that expressed an identifiable neoantigen and contained T cells that were reactive to that neoantigen. When CISH KO TILs were cultured with autologous DCs loaded with the neoantigen, more cells produced IFNγ, TNFα, and/or IL-2 than their WT counterparts. This was maintained even when low levels of the neoantigen were loaded, suggesting that CISH KO enhances both T cell functionality and sensitivity to antigenic stimulation. Similar results were observed using samples from a second patient, suggesting reproducibility.
Having shown that CISH KO could enhance the sensitivity of a T cell response to a neoantigen, the researchers wanted to know whether CISH KO could resurrect the functionality of neoantigen-specific T cells that had lost their ability to respond following REP. Using samples from a patient with TILs that were reactive to a neoantigen, but lost their reactivity after a REP, they found that while control TILs showed no reactivity to the neoantigen following REP, CISH KO TILs responded to neoantigen peptide-loaded DCs. This suggested that the loss of reactivity after traditional TIL REP could be restored with CISH KO.
Next, Palmer and Webber et al. wondered whether CISH might play a role in dampening immune responses to common neoantigens, like those that arise from TP53 driver mutations. Using 18 tumor samples with TP53 mutations and mutant p53 neoantigens, 7 of which showed evidence of T cell reactivity, the researchers found that genes associated with T cell activation were expressed in the reactive samples, while CISH expression clustered with non-reactive samples. When CISH was knocked out in a sample of TILs from a patient with a modest response to mutant p53, CISH KO TILs dramatically increased expression of IFNγ in response to mutant p53-loaded DCs, and more of the cells were polyfunctional compared to the unaltered TILs.
Interestingly, while CISH KO cells showed evidence of hyperactivation, they did not show signs of accelerated maturation, which would typically be expected. Investigating this unusual finding, the researchers found that PLC-γ1 and SLP76, intermediate molecules involved in TCR signaling, were phosphorylated with increased intensity and duration after TCR stimulation in CISH KO cells. Subsequently, there was an increase in the amplitude and duration of NFAT and NFκB translocation to the nucleus, a slight increase in c-Jun, and enhanced phosphorylation of ERK. However, enhanced phosphorylation of AKT, which is largely associated with T cell maturation, was not observed.
Given that CISH expression and PD-1 expression were initially found to be mutually exclusive, Palmer and Webber et al. evaluated whether CISH plays a role in the regulation of exhaustion markers and found that low CISH expression was associated with increased PD-1. In mice with gp-100-expressing melanoma tumors treated with adoptively transferred WT or CISH KO gp-100-specific pmel-1 cells, CISH KO cells were found to express higher levels of PD-1 eight days after adoptive transfer, suggesting potential synergy with PD-1 blockade. Comparing treatment combinations, the researchers found that while CISH KO pmel-1 cells alone or WT pmel-1 cells plus anti-PD-1 slowed tumor growth, CISH KO pmel-1 cells plus anti-PD-1 resulted in profound tumor regression and long-term survival of mice.
Together these results showed that CISH acts as an internal regulator of T cell reactivity, dampening responsiveness to neoantigens. CISH KO increased T cell sensitivity to neoantigens and enhanced their activation, functional responses, and tumor-killing capacity. CISH KO could potentially be used to enhance TIL therapy or improve responses to PD-1 checkpoint blockade, and warrants further study as a target for immunotherapy.
by Lauren Hitchings
Meet the researcher
This week, we asked first co-author Douglas Palmer to answer the following questions.

What prompted you to tackle this research question?
Understanding the role of negative regulators in T cell immunity has long been a passion of mine [1]. It began when we noticed that T cells acutely stop killing tumors after adoptive cell transfer [2] and I wanted to understand why and how we could reverse it. Unfortunately, at the time, genetic manipulation of these negative regulators in fully mature T cells was in its infancy and I was relegated to evaluating their role in tumor immunity using mouse knockout models and siRNA. What I uncovered though, was that the disruption of the Suppressor of Cytokine Signaling (SOCS) member, CISH, could result in profound and long-lasting tumor-free survival in animal models [3]. Surprisingly, I uncovered that this molecule blocked T cell receptor signaling and not cytokine signaling, as was previously thought at the time.
It is known that tumor-resident T cells (TIL) have neoantigen specificity for the tumors they reside in, however the tumor still grows, and the adoptive transfer of these cells fails to reliably result in tumor regression. I wanted to know how CISH related to other known markers of activation/exhaustion and if the deletion in fully mature TILs would restore their tumor killing capabilities. The advent of CRISPR and chemically-modified guide RNAs [4] was the big game changer that made this study possible.
What was the most surprising finding of this study for you?
What was most surprising was that in TILs, I found that CISH was inversely expressed with known activation/exhaustion markers like TOX, CD39, and PD-1. It appeared that CISH might inhibit their expression and the knockout of CISH resulted in upregulation of PD-1, which I exploited using a combinatorial approach.
What was the coolest thing you’ve learned (about) recently outside of work?
I’m an avid sailor and love to race. A few years back I took part in an ocean race from Annapolis, MD to St. George, Bermuda. There were about 40 other boats and I was crewing on a 50 ft sloop with 6 other guys. Our crew of seven was a motley bunch, all different demeanors and skill sets. One guy, an ex-Navy fighter pilot, was an excellent navigator. Another was a structural engineer and excellent at repairing broken sails. My specialty was sail aerodynamics, in particular spinnakers, which I trimmed for days on end. After we left the shelter of the Chesapeake Bay and entered the ocean, it was just the seven of us and we never saw another boat up until we got the reefs on the north shore of Bermuda six days later. Teamwork was key. Teamwork, getting along, and playing to our strengths got us through several big storms, ocean swells and eddies, and broken equipment. Teamwork got us safely to Bermuda and ultimately second place on the ocean portion of the race. I think understanding that concept will not only help get you across an ocean in a floating tub of plastic, but is central in developing new therapies to improve the lives of cancer patients.
1. Palmer, D.C. and N.P. Restifo, Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol, 2009. 30(12): p. 592-602.
2. Palmer, D.C., et al., Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci U S A, 2008. 105(23): p. 8061-6.
3. Palmer, D.C., et al., Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J Exp Med, 2015. 212(12): p. 2095-113.
4. Hendel, A., et al., Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology, 2015. 33(9): p. 985-989.



