Understanding how and why tumors become resistant to immunotherapy is one of the key obstacles in expanding the portion of patients who achieve a durable benefit from treatment. While some cancers show obvious mutations or genetic loss of antigen presentation, most cases of cancer relapse lack a clear mechanism of resistance. In two patients with Merkel cell carcinoma (MCC), Paulson et al. identified that transcriptomic changes induced by intense selective pressure may be to blame for tumor relapse following combination therapy.
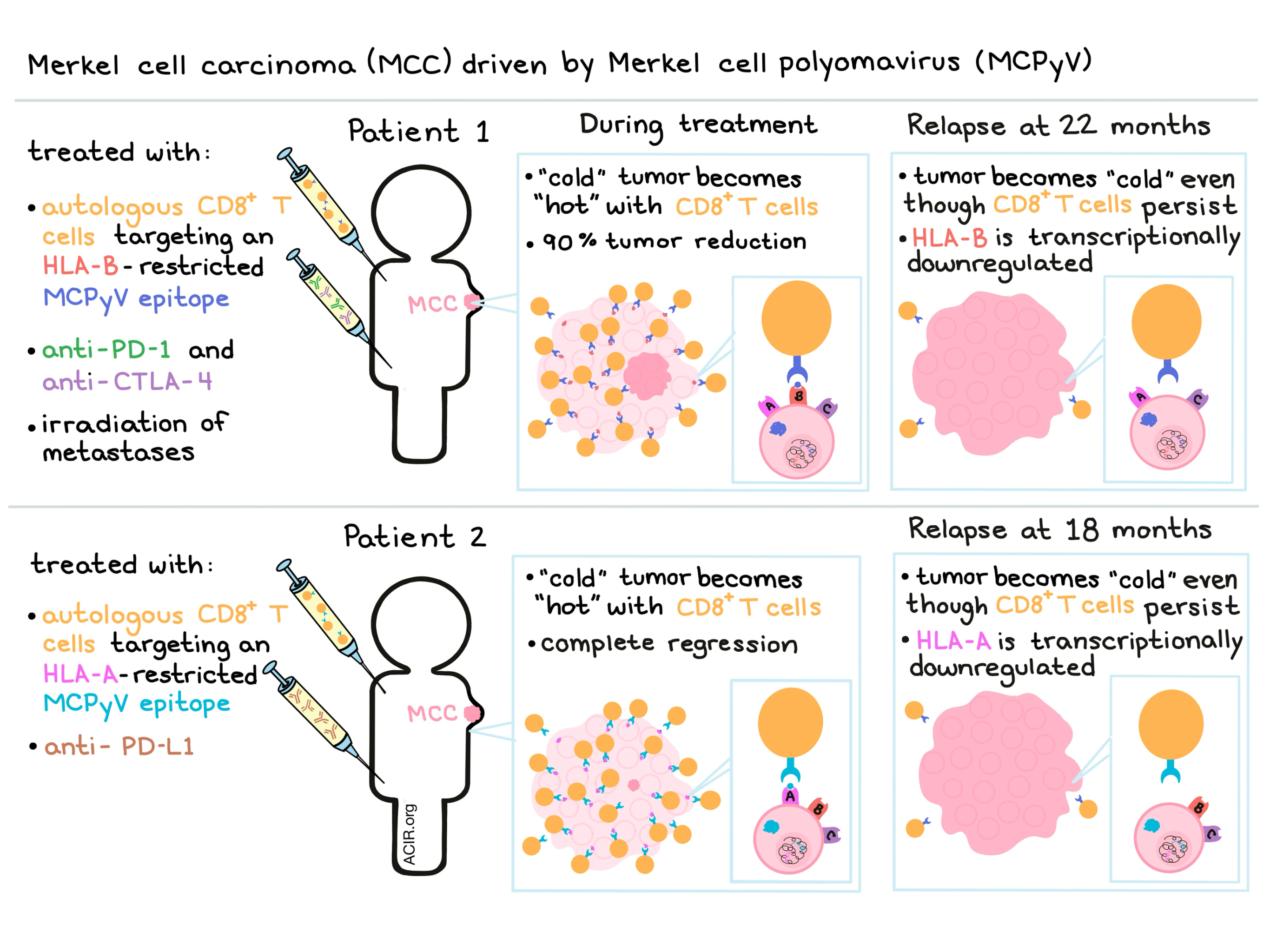
Paulson et al. began their study with a single patient with Merkel cell polyomavirus (MCPyV)-associated metastatic MCC. They identified a novel HLA-B-restricted epitope in MCPyV sT-Ag oncoprotein, and treated the patient with autologous, ex vivo-expanded CD8+ T cells specific for the epitope. Following adoptive T cell transfer (two infusions over 33 days), tumors continued to expand, and the patient was treated with pembrolizumab (anti-PD-1) and ipilimumab (anti-CTLA-4). The patient demonstrated dramatic symptomatic improvement and over 90% tumor reduction over the following year. After 22 months, however, the patient relapsed.
In an effort to identify the cause of relapse, Paulson et al. turned to the usual suspects, but found that none were to blame. Infused CD8+ T cells persisted in the peripheral blood, expression of HLA-ABC (detected with a pan anti-HLA antibody) was conserved, MCPyV oncoproteins were still expressed by the tumor, no mutations or loss of heterozygosity were observed in the HLA-B gene or promoter, and no new mutations were identified that would explain the acquired resistance. With no genetic mechanism to blame, the researchers evaluated whether transcriptional regulation played a role in the mechanism of escape.
Using single cell RNA sequencing (scRNAseq) and unsupervised clustering, the researchers found that activated effector T cells were significantly enriched at the time of response. Quantitative immunohistochemistry with HLA-DR (a marker of activated CD8+ T cells) revealed that activated CD8+ T cells highly infiltrated the shrinking tumors and were likely the primary mediators of tumor regression. At the time of relapse, however, T cell infiltration was low, indicating that the target antigen was likely undetectable to the T cells.
The transcriptomes of non-tumor cells before and after acquired resistance were nearly identical, indicating that a change in the tumor milieu was not likely the cause of relapse. scRNAseq of MCC tumor cells, however, demonstrated significant transcriptional changes between pre-and post-resistance biopsies, including significant downregulation of the HLA-B locus, but not HLA-A locus. Together, this data suggests that selective pressure from the infused HLA-B-restricted CD8+ T cells led to transcriptional changes that allowed for tumor immune escape. In short-term ex vivo cultures of the patient’s MCC, the researchers were able to reverse the downregulation of HLA-B using either pharmacologic doses of IFNγ or hypomethylating agent 5-azacitidine. The reversibility of the HLA-B downregulation is consistent with a mechanism of transcriptional rather than genetic HLA-B suppression.
To validate the results they had observed in the initial patient, Paulson et al. performed a similar series of analyses on a second patient. Like the first patient, the second patient was treated with autologous, ex vivo-expanded, MCPyV-specific T cells, however, in this patient the MCPyV-specific T cells infused were restricted to HLA-A rather than HLA-B, and the patient was treated with avelumab (anti-PD-L1) checkpoint inhibition rather than anti-PD-1 and anti-CTLA-4. The patient’s tumor went from immunologically cold to hot by day 14 and a complete response was achieved by 6 months, but after 18 months of ongoing treatment, the patient experienced a relapse that could not be attributed to lack of persistence of infused T cell product nor to any genetic mechanisms. Instead, the researchers observed selective transcriptional downregulation of the targeted HLA-A, but not HLA-B in the tumor cells. The researchers were unable to establish ex vivo cultures of this patient’s tumor cells, and therefore were unable to confirm whether the downregulation of HLA-A was reversible with drugs.
Together, the results from the initial and the validation patients show that tumors may utilize transcriptional downregulation of the specific HLA that is restricting a targeted epitope as a mechanism of immune escape. However, it is not yet clear whether this acquired capacity was caused by selective growth of a pre-existing tumor cell with the HLA transcriptionally suppressed, or whether the transcriptional suppression developed after treatment with multiple immunotherapeutic agents. Further research is required to better understand transcriptional downregulation of a specific HLA and the histologic tumor types it occurs in, to explore how often this mechanism is the cause of tumor escape, and to identify drugs and approaches to clinically reverse this phenomenon.
by Lauren Hitchings



